


Vol 100, No 5 (2023)

- Year: 2023
- Published: 22.11.2023
- Articles: 12
- URL: https://microbiol.crie.ru/jour/issue/view/180

ORIGINAL RESEARCHES
Studying the genetic diversity of the varicella-zoster virus in selected regions of the Russian Federation using high-throughput sequencing
Abstract
Introduction. Varicella-zoster virus (VZV), the causative agent of the disease of the same name and herpes zoster, is phylogenetically divided into 8 clades, the distribution of which is characterized by geographic reference to certain regions of the world. For most countries, VZV clades circulating in their territories have been identified, however, such information is almost unavailable for Russia.
The purpose of the study is to develop an effective method for VZV typing using high-throughput sequencing technologies to identify the prevalence of various VZV clades in Moscow, Moscow Region, and Stavropol Territory.
Materials and methods. To genotype VZV, it is enough to refer to 7 nucleotide positions. Their unique combinations can be used to assign the virus to one of the clades. Short sections of nucleotide sequences of open reading frames were obtained using a developed set of primers.
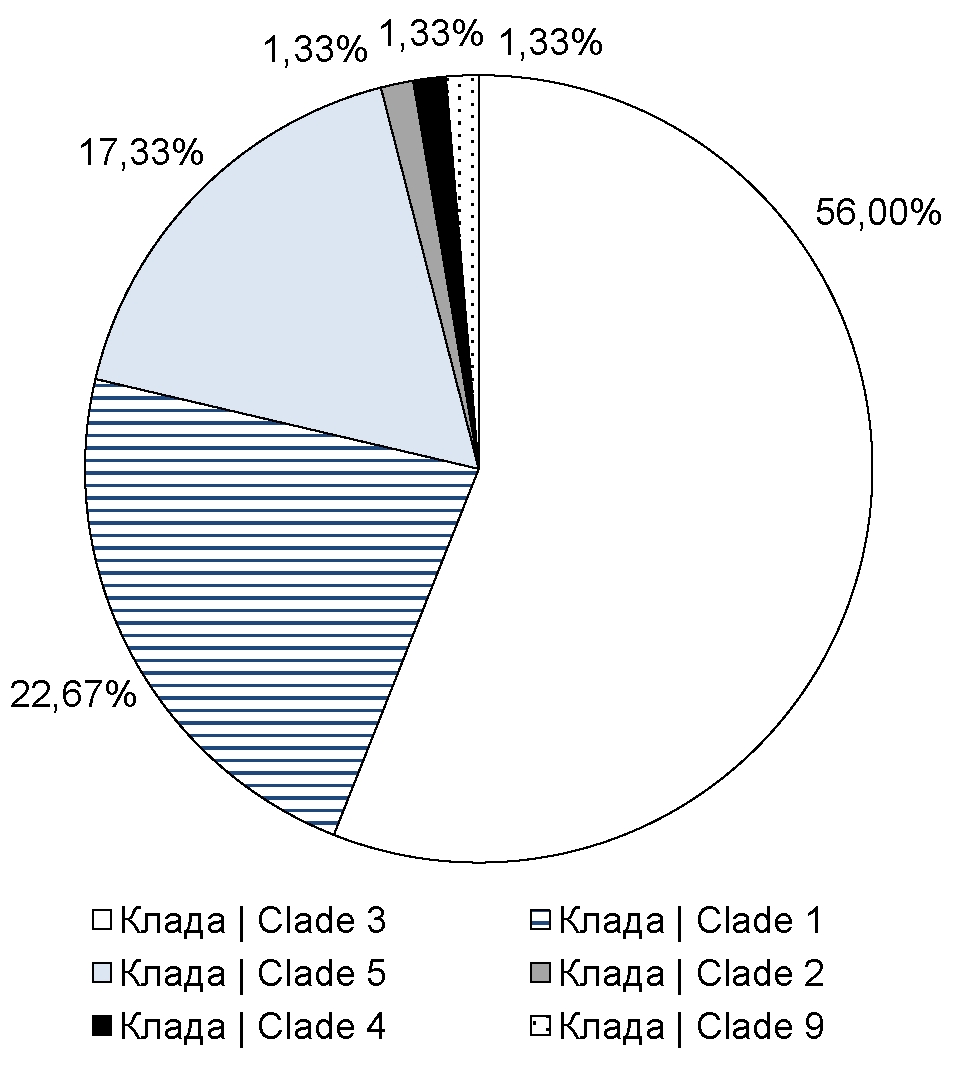
Results. A VZV genotyping technique has been developed and optimized. Using this technique, primary data on the distribution of VZV clades in the studied regions have been obtained. Thus, it has been established that in Moscow and a number of other regions, the 1st, 3rd, and 5th clades of VZV are predominantly distributed.
Conclusion. The developed technique, including a primer panel and a genotyping algorithm, allows VZV typing in a short time while reducing specimen preparation costs and simultaneously increasing the number of specimens in one sequencing cycle. The results obtained using this assay allow us to assume that in Moscow, Moscow Region, Stavropol Territory, VZV, clades 1, 3, and 5 are the most represented ones. To confirm this hypothesis, it is necessary to include a larger number of clinical specimens in subsequent studies, including from other regions of the country.
 267-275
267-275



Analysis of production levels of InlA and InlB invasion factors in Listeria monocytogenes isolates collected in the Russian Federation
Abstract
Background. Listeria monocytogenes is characterized by the presence of epidemic hypervirulent clones. A key feature of L. monocytogenes is its capacity to invade non-professional phagocytic cells. Hypervirulent clones are strongly associated with the increased production and/or the presence of certain isoforms of invasion factors InlA and InlB.
The purpose of the study is to create a test system for InlA and InlB detection and to measure the InlA and InlB production levels in L. monocytogenes isolates belonging to clonal groups with different virulence potential.
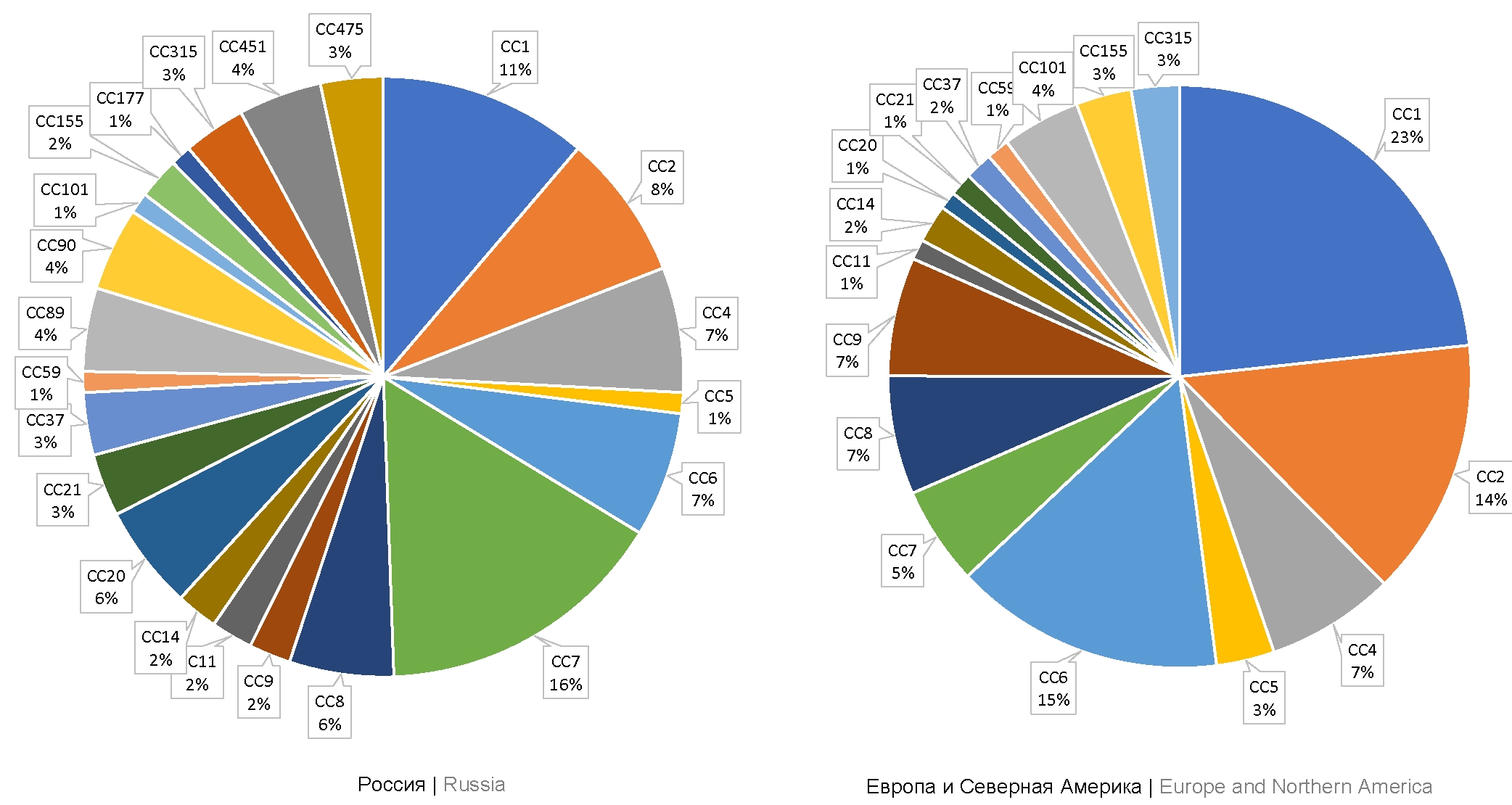
Materials and methods. The study was performed using 32 L. monocytogenes strains belonging to epidemic clones ECII, ECIV, ECVII (clonal complexes CC1, CC2, CC7) and hypovirulent clonal complex CC9. Sequencing of inlA and inlB genes was performed. The indirect enzyme-linked immunosorbent assay was used to analyze the production levels of InlA and InlB proteins.
Results. The variability of InlA was revealed among strains belonging to the same clonal complex: 3 InlA isoforms were identified among strains belonging to CC7; out of 8 strains belonging to CC9, one strain had a stop codon in the inlA gene, leading to the loss of function of the InlA protein. The differences between inlB alleles correlated with the specificity of strains belonging to a certain clonal complex. Differences in production levels of invasion factors were measured. In strains belonging to CC9, the InlA production level was 2.5 times as low compared to strains belonging to CC1, CC2, and CC7. In strains belonging to phylogenetically related CC1 and CC2, the InlB production level was on average 4 times as high compared to strains belonging to CC7 and CC9.
Conclusion. The obtained results confirm the variability of major invasion factors both among clonal complexes and strains of the same complex. The increased production of invasion factors InlA and InlB correlates with the potential virulence of strains.
276-286
Characteristics of antibiotic resistance of non-typhoidal Salmonella circulating in the Russian Federation in the period from 2019 to 2022
Abstract
Introduction. Non-typhoidal Salmonella make a significant contribution to the incidence of enteric infections and are characterized by an increasing proportion of strains resistant to antimicrobial agents (AMA), including the first choice antibiotics (cephalosporins III and fluoroquinolones).
The purpose of the study is to assess the phenotypic resistance of Salmonella to various classes of AMAs and determine the relationship between the phenotypic resistance, serotype, source of isolation and nature of incidence.
Materials and methods. We studied 752 representative strains of Salmonella of 2494 strains isolated from various sources (clinical samples, food products, environment) received from 59 regions of Russia in the period from 2019 to 2022. The phenotypic resistance to 22 antibiotics of 11 CLSI classes of AMAs was assessed by broth microdilution method (minimum inhibitory concentration). The diversity of resistance profiles of Salmonella serotypes was compared using the Shannon index.
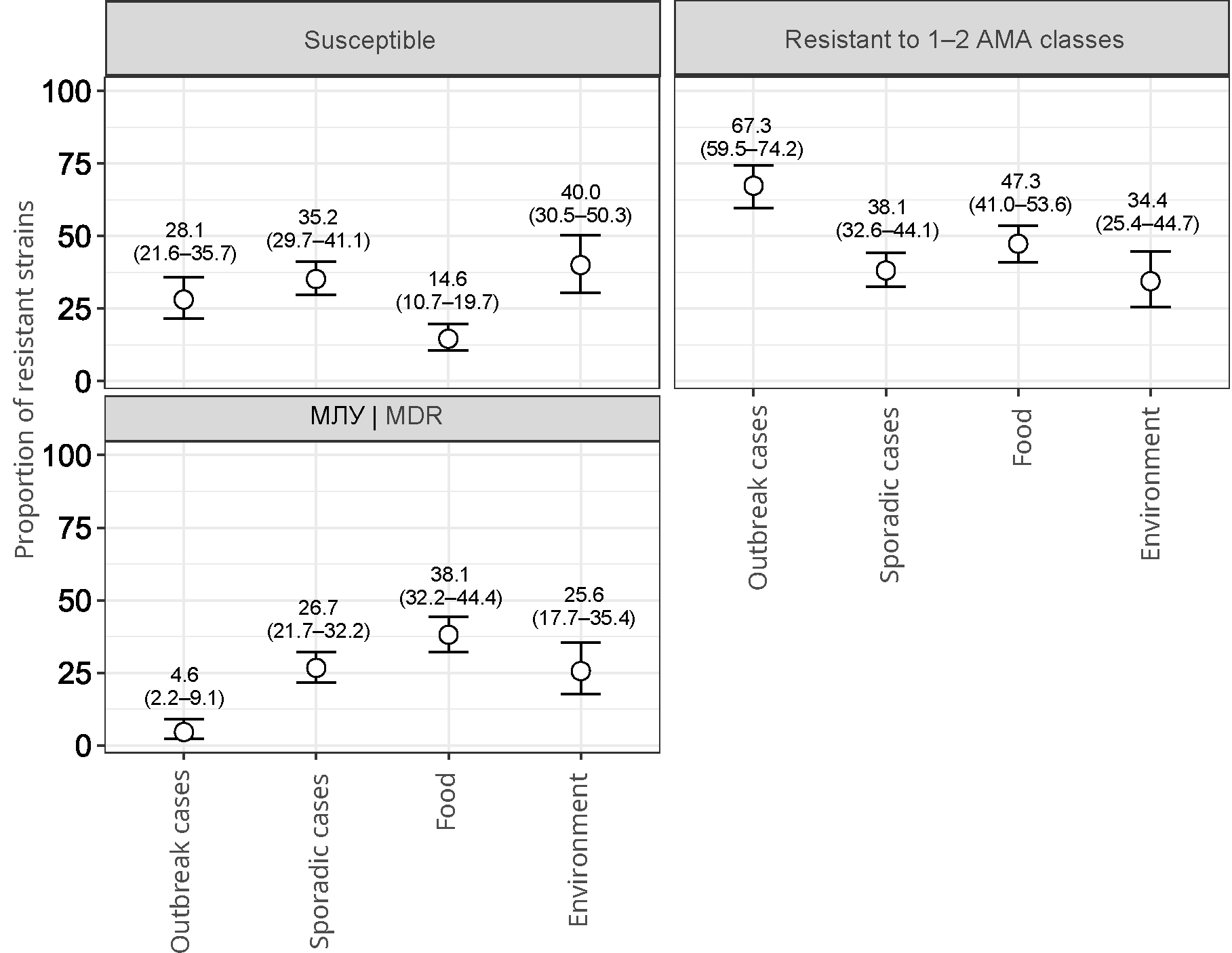
Results. The dominant position in terms of isolation frequency is occupied by the serotypes Salmonella Enteritidis, S. Infantis, S. Muenchen, S. Typhimurium, S. Bovismorbificans, which accounted for 64.4% of the studied strains. 543 (72.2%) strains showed resistance to at least one of the tested antibiotics; 193 (25.7%) strains were characterized by multidrug resistance phenotype (MDR). Resistance to AMA classes was characterized by the following distribution: quinolones (61.3%), tetracyclines (28.1%), penicillins (19.1%), β-lactam combination agents (18.6%), folate pathway antagonists (16, 5%), phenicols (10.1%), aminoglycosides (5.6%), cephems (4.7%), monobactams (4.4%), lipopeptides (3.9%). No penem-resistant strains have been identified. The features of Salmonella resistance by AMA classes are shown to depend on the sources of isolation, the Salmonella serotype and the nature of the incidence (outbreak and sporadic).
Conclusions. Monitoring of phenotypic antibiotic resistance is an important tool for epidemiological surveillance in order to prevent the spread of bacterial resistance to AMAs.
287-301
Comparative evaluation of disinfectant efficacy against biofilm-residing microorganisms
Abstract
Introduction. Bacteria in biofilms (BFs) have increased resistance to antibacterial agents, including disinfectants; however, the efficacy level varies depending on the chosen treatment. Therefore, evaluation of efficacy of main disinfectants against BF-residing microorganisms is of scientific and practical interest.
The purpose of the study was to explore the effect of disinfectants from various chemical groups on gram-positive and gram-negative bacteria residing in BFs.
Materials and methods. The effect of the following disinfectants has been evaluated: alkyldimethylbenzylammonium chloride (ADBAC), tertiary amine (TA), polyhexamethylene guanidine chloride (PHMG), hydrogen peroxide (HP), chloramine (CA), dichloroisocyanuric acid sodium salt (Na DCC), sodium hypochlorite (HC), ethyl alcohol (EA), glutaraldehyde (GA)) against Pseudomonas aeruginosa ATCC 15442 and Staphylococcus aureus ATCC 6538-P BFs. BFs were grown in 96-well plates at 37ºC for 24 hours and then exposed to biocide solutions. The efficacy of disinfectants was evaluated by the number of remaining viable cells and BF relative density.
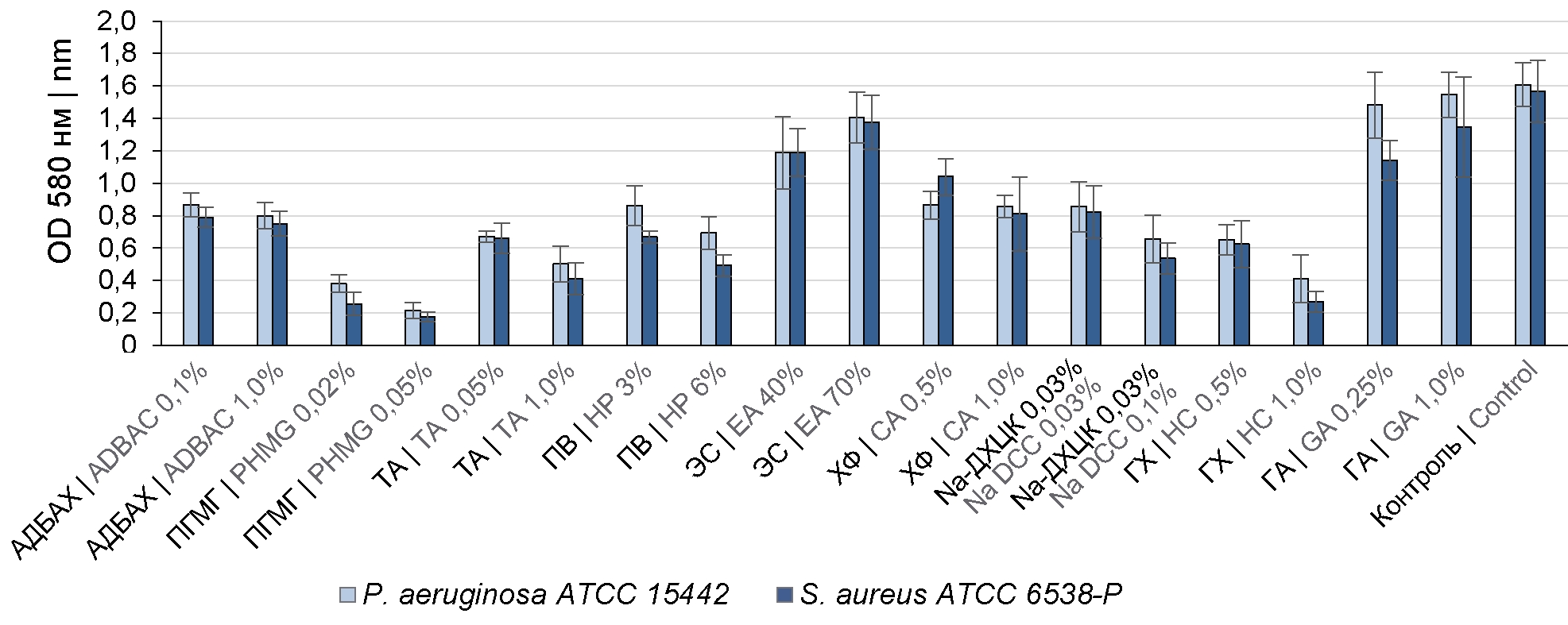
Results. The analyzed bacterial strains formed moderate BFs; the average number of viable cells in BFs was 6.51 ± 0.19 lg. The viable bacterial cell counts in BFs reduced by more than 4 lg when exposed to HP solutions at a concentration of 6%, Na DCC solution — 0.1% (by active chlorine), HC — 1% (by active chlorine), CA – 1% (by product), PHMG — 0.05%, TA — 1.0 %. The BF density decreased by more than 70%. ADBAC solutions at concentrations of 0.1–1.0%, TA — 0.05%, HP — 3%, Na DCC solution — 0.05% (by active chlorine) caused a 2-lg reduction in viable cell counts in BFs. The efficacy of chlorine-active compounds and HP increased when 0.5% sulfonol was added. GA (0.25–1.00%) and EA (40–70%) solutions were ineffective against BF microorganisms.
Conclusion. A promising potential in combating microbial biofilms is demonstrated by disinfectants from the group of oxidizing agents (chlorine-active and oxygen-containing), TA and PHMG; using ADBAC as an individual compound is ineffective; aldehydes and alcohols are unable to destroy BFs and eliminate microorganisms in them.
302-309
Isolation and genetic analysis of the chikungunya virus from Aedes aegypti and Aedes albopictus mosquitoes captured in Central America
Abstract
Introduction. The habitat of mosquitoes belonging to the genera Aedes spp., Culex spp., Culiseta spp. is in South and Central America, including Nicaragua. Monitoring of the spread of mosquito vectors and assessment of the infection with arboviruses can provide information on possible occurrence of new diseases or an increase in the reported cases, changes in the infectivity of viruses for humans due to changes in pathogen transmitters.
The purpose of this study was isolation and identification of arboviruses belonging to the Flavivirus and Alphavirus genera from A. albopictus, A. aegypti, Culiseta spp., Culex spp. mosquitoes captured in forests of Nicaragua.
Materials and methods. A. albopictus, A. aegypti, Culiseta spp., Culex spp. mosquitoes were captured during the dry season in 2021 in forested areas of Nicaragua in four different locations. Mosquitoes were sorted into pools, each containing 5-8 mosquitoes (236 pools in total). Using the reverse transcription polymerase chain reaction, the pools were tested for the presence of chikungunya (CHIKV), dengue, Zika, and yellow fever viruses. Positive pools were inoculated into the C6/36 cell culture to obtain isolates and for their further sequencing.
Results. The dengue virus was detected only in Aedes spp. mosquitoes: in 7 pools — A. aegypti, in 1 — A. albopictus. CHIKV was also detected only in Aedes spp. mosquitoes: in 3 pools — A. aegypti, in 1 — A. albopictus. The sequencing of nucleotide sequences of 6К, Е1, Е2, and NS1 genes of CHIKV isolated from A. albopictus mosquitoes showed that compared to the similar gene sequences from CHIKV isolates recovered from A. aegypti mosquitoes, the 6K gene region contained 4 nucleotide and 4 amino acid substitutions, while the E1 region contained 16 nucleotide substitutions, 10 of them led to amino acid substitutions; the E2 region contained 14 nucleotide and 11 amino acid substitutions; the NS1 region contained 33 nucleotide and 19 amino acid substitutions.
310-318
Comparative analysis of IgG levels in blood sera from patients with COVID-19, persons vaccinated by «Gam-COVID-Vac» and healthy donors before the pandemic
Abstract
Introduction. Serum IgG measurement is used in the diagnosis and monitoring of many diseases. Therefore, it seems important to study the impact of coronavirus pandemic on IgG levels in population and the role of this parameter in COVID-19.
The aim of this study was to compare mean IgG levels in sera obtained from 31 hospitalized COVID-19 patients, 30 healthy donors before pandemic and 34 donors vaccinated with «Sputnik V» (have not had COVID-19).
Materials and methods. Total IgG was quantitated by two enzyme immunoassays (EIAs): «IgG Total-ELISA-BEST» kit certificated in Russia and homemade competitive EIA utilizing bispecific monoclonal antibodies (bsAbs) against human IgG (HIgG) and horseradish peroxidase (HRP).
Results and discussion. The groups did not show differences in IgG levels (regardless of sex) with both methods giving comparable results. However, «IgG Total-ELISA-BEST» kit revealed statistically significant differences in mean serum IgG levels in subgroups of male patients depending on the levels of antibodies to viral RBD-antigen: below and above 400 BAU/ml. In the first subgroup (10 men) the mean serum IgG content was 14.3 ± 4.1 mg/mL, while in the second (6 men) — 6.9 ± 2.7 mg/mL.
Conclusion. Sera obtained before pandemic contained the same mean IgG concentrations as sera from donors vaccinated with «Sputnik V» and COVID-19 patients. The relatively decreased mean IgG concentration was found only in COVID-19 male patients with anti-RBD antibodies levels above 400 BAU/ml. In light of literature data on association of decreased serum IgG with COVID-19 severity, it would be reasonable to further compare larger groups, taking into account clinical differences. The possibility of using bsAbs for human Ig measurements by competitive EIA was demonstrated.
319-327
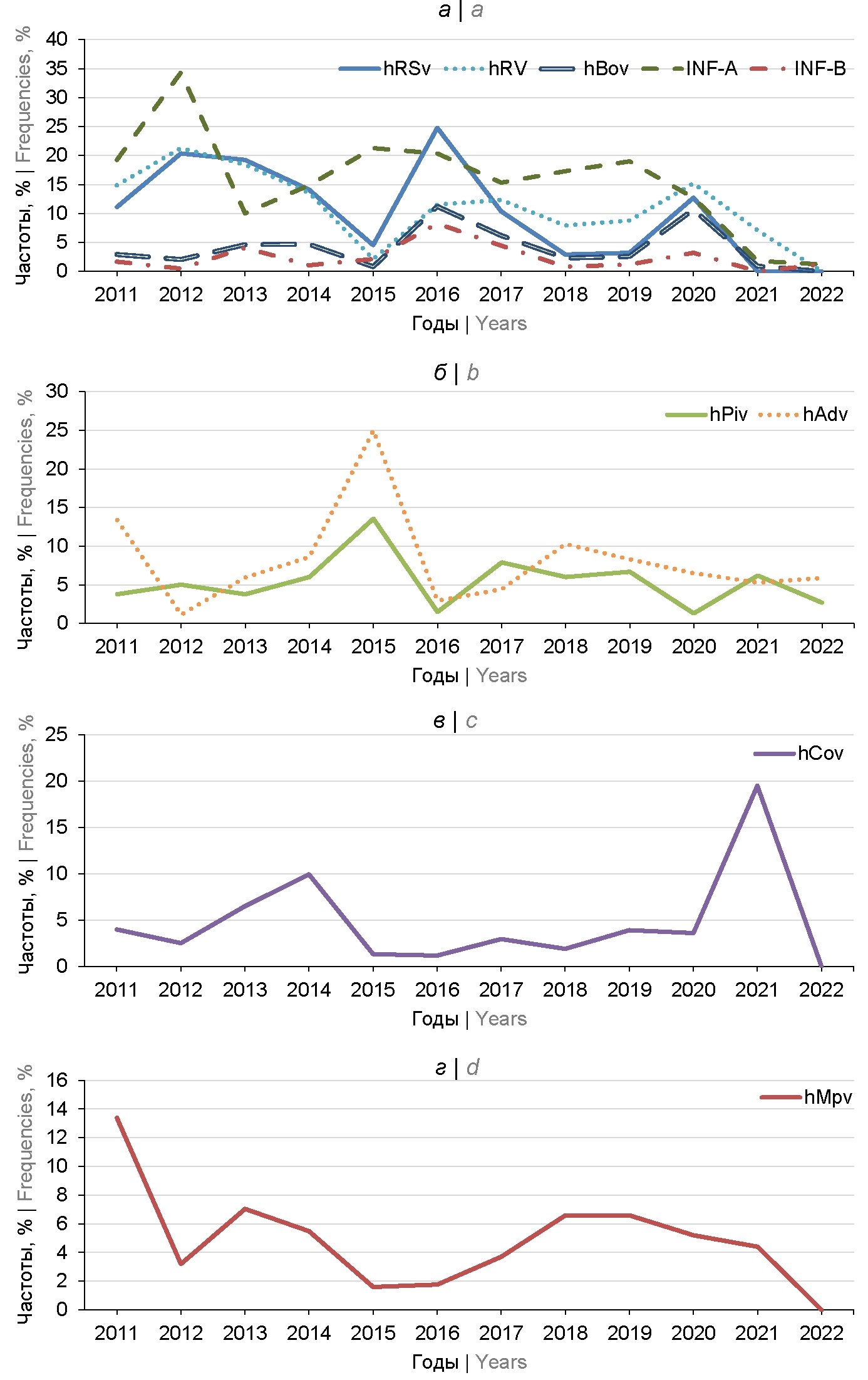
Monitoring of respiratory viral infections in Moscow during 2011–2022
Abstract
Introduction. Respiratory viruses (RV) circulate throughout the world and in all seasons of the year. Long-term monitoring of the distribution of respiratory pathogens is necessary to analyze the relevance of diagnostic systems to current viral isolates, to assess the risks of infection and the need for vaccine development and use, as well as to investigate the interdependence of RV reproduction in mixed infections.
Objective — to study the causative agents of acute respiratory viral infections (ARVI) in Moscow during 2011–2022 by reverse transcription with subsequent polymerase chain reaction with fluorescent hydrolysis probes detection in real-time (RT2-PCR).
Materials and Methods. Nasopharyngeal swabs from 3908 patients with acute respiratory infections were examined by the RT2-PCR.
Results. Monitoring of RV spread in Moscow showed cyclical changes in frequencies with three dominant species: influenza A virus (up to 31.3%), respiratory syncytial virus (up to 24.8%) and human rhinoviruses (up to 21.3%) in 2011–2020. The increase in the portion of unidentified clinical specimens from 1.2 to 28.5% in 2022 indicated incomplete accordance of diagnostic systems to modern RV isolates or the emergence of new species or strains of pathogens. Unidirectional changes in dynamics were registered for 5 out of 9 studied RVs with correlation coefficients of 0.43–0.79. High frequencies of mixed acute respiratory viral infections (up to 33.4%) along with unidentified samples do not allow us to accurately assess the risks of infection with various RV in Moscow, but prove the necessity of preventing infectious diseases with the most common RV.
Conclusion. Analysis of the dynamics of RV frequencies in Moscow showed the preservation of the dominant species: influenza A virus, respiratory syncytial virus and human rhinoviruses. During the period of vaccination against COVID-19, the proportion of seasonal coronaviruses increased.
328-337
Molecular epidemiological monitoring of the tuberculosis pathogen in the Arkhangelsk region
Abstract
Introduction. Against the background of improvement of the main epidemiological indicators (morbidity and mortality) for tuberculosis in the Arkhangelsk region, the proportion of newly diagnosed tuberculosis patients with multidrug-resistant pathogen (MDR-TB) increased from 18.7% in 2002 to 33.8% in 2018.
The purpose of this study was the genotypic characterization of Mycobacterium tuberculosis strains obtained from newly diagnosed tuberculosis patients in the Arkhangelsk region in 2018.
Materials and methods. 89 M. tuberculosis strains isolated in 2018 from newly diagnosed tuberculosis patients were studied. Beijing genotype, its clusters B0/W148 and Central Asian/Russian were determined by PCR detection of the specific markers: IS6110 insertions in the dnaA-dnaN region, mutations in codons 48 of the mutT4 gene (CGG > GGG) and 58 of the mutT2 gene (GGA > CGA), IS6110 insertions in the Rv2664 region-Rv2665 and Rv1359-Rv1360, substitutions G > A in the sigE gene. Non-Beijing strains were spoligotyped.
Results. Drug resistance was detected in 41.6% (37/89), MDR — in 33.7% of strains. In 90% (27/30) of MDR strains, resistance to rifampicin and isoniazid was due to rpoB Ser531Leu and katG Ser315Thr mutations. Following M. tuberculosis genotypes were identified: Beijing (67.4%), T (14.6%), Ural (4.5%), Haarlem (4.5%), LAM (2.3%) and CAS1-Delhi (1.1%). Among the Beijing strains, clusters Central-Asian/Russian (60%; 36/60) and B0/W148 (30%; 18/60) prevailed. The majority of MDR strains belonged to the Beijing family (93.3%; 28/30), of which 64.3% (18/28) and 21.4% (6/28) belonged to clusters B0/W148 and Central-Asian/Russian, respectively.
Conclusion. In heterogeneous population of the causative agent of tuberculosis in the Arkhangelsk region, the most common strains were those of the Beijing genotype; in 2018 its share increased to 67.4% (40.4% in 1998–1999). Among MDR strains, the proportion of Beijing reached 93.3%, of which more than half (64.3%) belonged to the epidemiologically and clinically significant in Russia cluster B0/W148.
338-345
Analysis of changes in the genome of Vibrio cholerae O1 El Tor genovariants during the current period of the cholera pandemic
Abstract
Introduction. The genome variability of genetic variants of El Tor cholera agent has led to the emergence of strains carrying mutations in various genes associated with epidemically important pathogen properties. This situation requires an assessment of the trends in these changes in order to predict the pathogenic potential of previously unknown variants and promptly develop new tools for their diagnostics and prevention.
The purpose of this work was to analyze the dynamic changes in pathogenicity and drug resistance genes of V. cholerae El Tor genetic variants from endemic countries and Russia.
Materials and methods. We analyzed complete genome nucleotide sequences of 104 V. cholerae El Tor strains from the NCBI Gen Bank and European Nucleotide Archive databases, as well as those obtained by us. The nucleotide sequences were analyzed using the UGEN v. 45.1 software. The dendrogram was constructed using maximum parsimony algorithm in BioNumerics v.7.6 software package based on the multiple alignment generated using the Snippy 4.6.0 program.
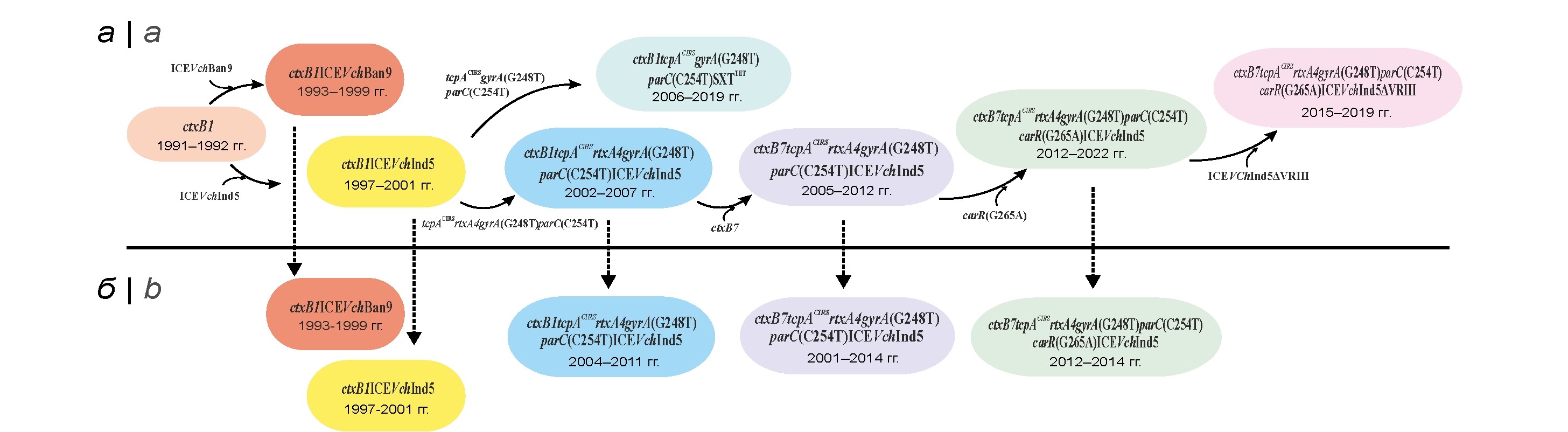
Results. Genomic sequences of 103 strain genovariants isolated on the territory of nine endemic countries of Asia and Africa, as well as in Russia in 1991-2022, have been compared. It is shown that the process of genovariant genome changing was multistage and occurred due to the continuous accumulation of point mutations in key (ctxB and tcpA) and additional (rtxA) genes of pathogenicity and core genes of antibiotic resistance (gyrA, parC and carR), as well as a deletion in SXT element. The most important was the change in the ctxB gene and the emergence of new genovariants with the ctxB7 allele, which replaced the previously prevalent strains. Analysis of altered genome regions of 83 strains from endemic regions has revealed eight genotypes, while the strains (21 isolates) imported to Russia belonged to only five of them including highly virulent strains with the ctxB7 allele and lost PolR biovar-specific feature due to carR gene mutation. The established close phylogenetic relatedness of genovariants from Russia with strains from endemic Asian countries confirms their importation from this region.
Conclusion. The sequential occurrence and accumulation of mutations in the pathogenicity and drug resistance genes in the genome of genovariants in endemic regions have been shown, which leads to a change in their epidemically important features. The importation of new highly virulent genovariants into Russia has been established, which indicates the need for an ongoing assessment of changes in the genome of this pathogen for the timely development of adequate means of gene diagnostics and prevention.
346-357
Analysis of coxsackievirus A6 circulation in the territories of the Far Eastern Federal district of the Russian Federation in 2014–2019
Abstract
Introduction. Molecular epidemiological monitoring of enterovirus infection (EVI) in the territories of the Russian Federation showed that coxsackievirus A6 (CVA-6) had been one of the most prevalent types of enteroviruses that circulated among the country population during last years and had caused majority of EVI outbreaks.
Objective — to evaluate coxsackievirus A6 circulation in the territories of the Far Eastern Federal district (FEFD) in 2014–2019 utilizing methods of molecular genetics.
Materials and methods. RT-PCR was employed to detect enterovirus RNA in biological material collected in 9 territories of the FEFD . In order to establish enterovirus types, amplification of positive samples was carried out to detect nucleotide sequence fragments of the VP1 and VP2 genes. Molecular genetic analysis of the Far Eastern CVA-6 strains was based on detection of nucleotide sequences of VP1 and 3Dpol gene fragments. Phylogenetic trees were constructed by the means of Bayesian phylogenetic methods.
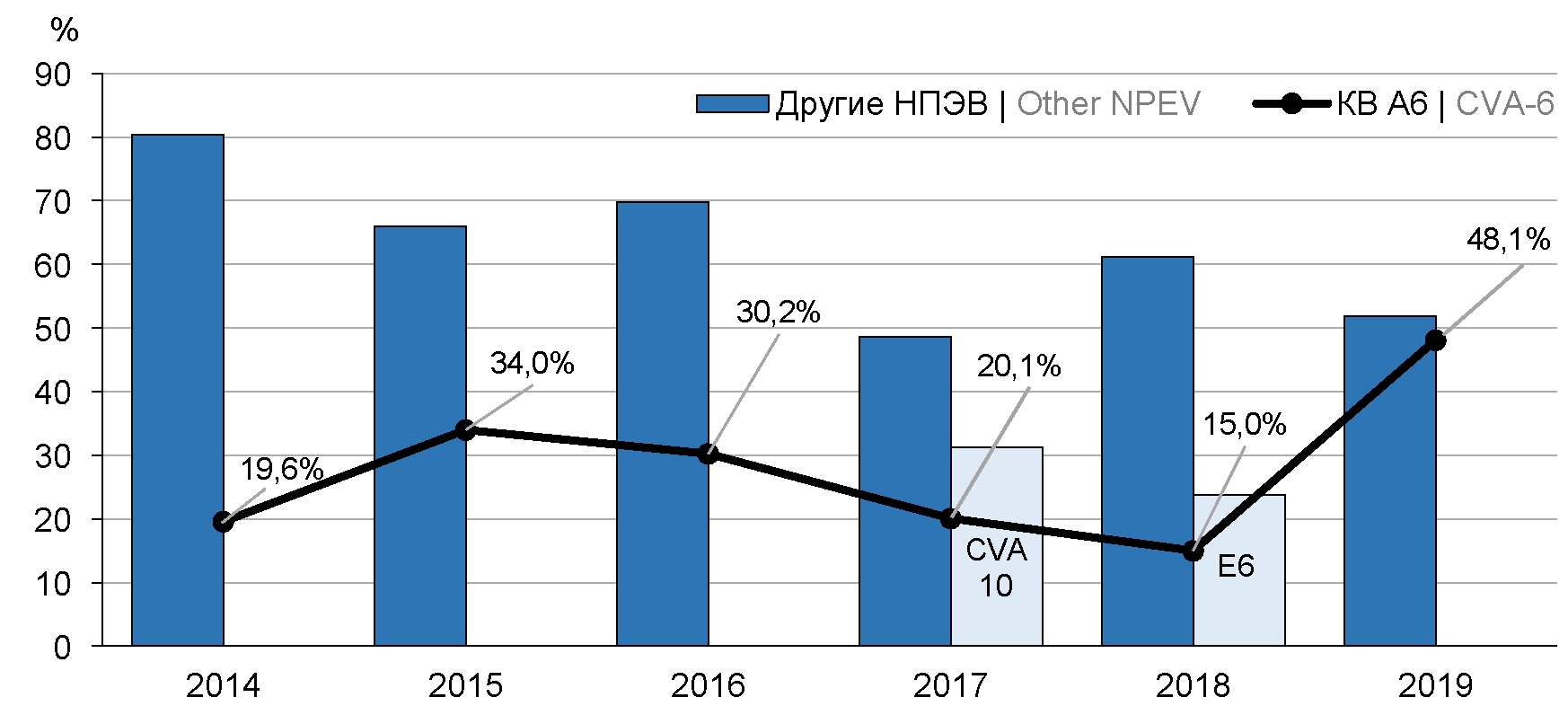
Results. Total 1773 nucleotide sequences of 43 types of non-polio enteroviruses were obtained in 2014-2019. Majority of the sequences belonged to coxsackievirus A6 (524; 29.5%). In the years of the highest CVA-6 detection an increase in EVI incidence as well as EVI outbreaks were observed in the territories of FEFD. The most prevalent manifestations of EVI caused by CVA-6 in FEFD were herpangina and exanthemic forms. Phylogenetic analysis showed that Far Eastern strains of CVA-6 during in the analyzed period of time belonged to D3 subgenotype that is dominant in the world. The circulation of several recombinant forms of CVA-6 (RF-A, -H, -L, -N, -R) was also registered.
Conclusion. The genetic diversity of CVA-6 strains circulating in the territories of the FEFD in 2014–2019 revealed in this study requires further investigation in order to obtain new knowledge about the CVA-6 molecular epidemiology and improve the enterovirus surveillance system.
358-368
Phylodynamic characteristics of the LMP-1 gene of the Epstein–Barr virus isolated in the Nizhny Novgorod region
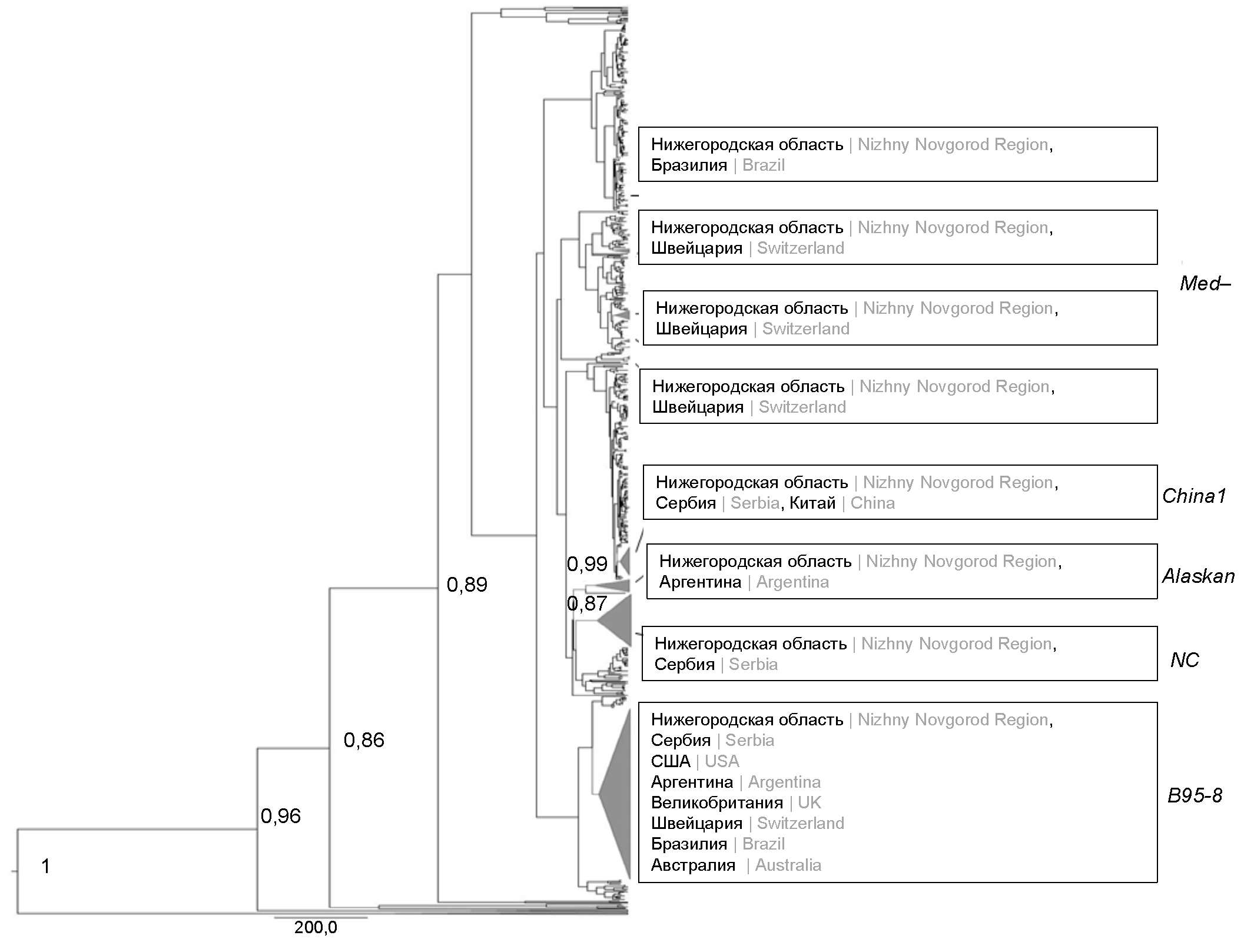
Abstract
Introduction. Epstein–Barr virus (EBV) is one of the most common herpesviruses and has a pronounced genetic polymorphism. The study of the phylodynamic characteristics of the virus is an important aspect of the study of evolutionary changes in the LMP-1 gene and their consequences.
The aim of the work was a philodynamic analysis of EBV isolates from Nizhny Novgorod region based on the C-terminal fragment of the LMP-1 gene.
Materials and methods. The study included 158 EBV isolates obtained from blood leukocytes and saliva of children aged 1–17 years with a diagnosis of infectious mononucleosis caused by EBV (n = 68) and apparently healthy children of comparable sex and age (n = 29). LMP-1 genovariants were obtained using the Sanger sequencing method. Comparative analysis of amino acid sequences was performed using the MEGA X program. Philodynamic analysis of the obtained nucleotide sequences and isolates deposited in GenBank was carried out using the BEAST v. 1.10.4 software package. Recombination analysis was performed using the Simplot program.
Results. 158 nucleotide sequences of the C-terminal fragment of the LMP-1 gene from Nizhny Novgorod region EBV isolates were obtained and deposited in the GenBank database. The circulation time of the nearest common ancestor for the modified B95-8 genovariants with G212S + E328Q + S366T and NC mutations with the D250N substitution has been established dating back to 1994 and 1923. The rate of evolution of these genovariants was the highest and amounted to 1.298 × 10–4 and 7.868 × 10–4 nucleotide substitutions/site/year. Recombinations were detected in the Nizhny Novgorod region sequences Med-, B95-8, China 1 with mutations G212S, G212S, E214Q, respectively.
Conclusion. For the first time, a phylodynamic characterization of Nizhny Novgorod region isolates and LMP-1 EBV genovariants isolated in various regions of the world is given. The data obtained expand the existing understanding of the circulation of EBV LMP-1 genovariants in the territory of the European part of Russia.
369-379
REVIEWS
Molecular-genetic portrait of virulence of Stenotrophomonas maltophilia
Abstract
Introduction. Stenotrophomonas maltophilia is an opportunistic pathogen that is intrinsically resistant to a wide range of antibiotics. The bacterium is associated with a number of serious diseases and makes a significant contribution to the pathogenesis of polymicrobial infections. S. maltophilia has a wide range of virulence factors, information about which is currently presented in the form of scattered and unconsolidated data.
Purposes and objectives: critically analyze and summarize current data regarding the molecular-genetic aspects of S. maltophilia virulence for better understanding of the pathogenesis of infections associated with this pathogen.
Materials and methods. An analysis of information from 80 modern literary sources devoted to the study of the virulent properties of S. maltophilia at the molecular-genetic level has been carried out. The analysis focuses on the mechanisms of production of virulence factors and their genetic determinants.
Results.The molecular mechanisms of virulence that determine the infectious process caused by S. maltophilia have been analyzed and summarized, including the adhesive function of the surface structures of the bacterial cell (lipopolysaccharides, pili/fimbriae, flagella), the production of extracellular enzymes, the ability to form biofilms on abiotic surfaces and on the tissues of the macroorganism, the functioning of efflux pumps, secretion of small molecules into the external environment by the intercellular information exchange system Quorum Sensing, as well as the influence of iron metabolism on the virulence properties of S. maltophilia.
Conclusion. The adaptation mechanisms that allow S. maltophilia to adapt to new habitat niches and survive in the human body and unfavorable environmental conditions have been poorly studied. An analytical review summarizing current information on the molecular-genetic aspects of S. maltophilia virulence will be of interest to clinicians and researchers studying the fundamental mechanisms of virulence.
380-390
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.