


Studying the genetic diversity of the varicella-zoster virus in selected regions of the Russian Federation using high-throughput sequencing
- Authors: Nadtoka M.I.1, Lysenkov V.G.1, Agletdinov M.R.1, Mishkin A.A.1, Afonina N.M.1, Ploskireva A.A.1, Mikheeva I.V.1, Khafizov K.F.1, Akimkin V.G.1
-
Affiliations:
- Central Research Institute of Epidemiology
- Issue: Vol 100, No 5 (2023)
- Pages: 267-275
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18473
- DOI: https://doi.org/10.36233/0372-9311-423
- EDN: https://elibrary.ru/tkklhp
- ID: 18473

Cite item

Abstract
Introduction. Varicella-zoster virus (VZV), the causative agent of the disease of the same name and herpes zoster, is phylogenetically divided into 8 clades, the distribution of which is characterized by geographic reference to certain regions of the world. For most countries, VZV clades circulating in their territories have been identified, however, such information is almost unavailable for Russia.
The purpose of the study is to develop an effective method for VZV typing using high-throughput sequencing technologies to identify the prevalence of various VZV clades in Moscow, Moscow Region, and Stavropol Territory.
Materials and methods. To genotype VZV, it is enough to refer to 7 nucleotide positions. Their unique combinations can be used to assign the virus to one of the clades. Short sections of nucleotide sequences of open reading frames were obtained using a developed set of primers.
Results. A VZV genotyping technique has been developed and optimized. Using this technique, primary data on the distribution of VZV clades in the studied regions have been obtained. Thus, it has been established that in Moscow and a number of other regions, the 1st, 3rd, and 5th clades of VZV are predominantly distributed.
Conclusion. The developed technique, including a primer panel and a genotyping algorithm, allows VZV typing in a short time while reducing specimen preparation costs and simultaneously increasing the number of specimens in one sequencing cycle. The results obtained using this assay allow us to assume that in Moscow, Moscow Region, Stavropol Territory, VZV, clades 1, 3, and 5 are the most represented ones. To confirm this hypothesis, it is necessary to include a larger number of clinical specimens in subsequent studies, including from other regions of the country.
Full Text
Introduction
Varicella-zoster virus (VZV) is a member of the Alphaherpesvirinae subfamily, genus Varicellovirus, of the family Herpesviridae. The characteristic properties of this subfamily representatives are a short reproductive cycle, rapid propagation, and effective destruction of infected cells. VZV is able to replicate in a limited range of host organisms, which includes only cells of human and simian origin. The annual incidence of varicella zoster (VZ) ranges from 13 to 16 cases per 1,000 people, with significant year-by-year variations. In countries with temperate climates, the incidence of VZ, depending on age, is the highest among children of preschool (1–4 years) or primary school age (5–9 years) and amounts to more than 100 cases per 1,000 children a year. Thus, more than 90% of people become infected before adolescence, and only a small proportion of adults (<5–10%) remain susceptible to the disease. In the tropical climate, the exposure to infection occurs on average at an older age, with a higher proportion of infected cases among adults [1, 2].
VZV infection has a pronounced seasonal nature, with peak incidence occurring in winter and spring, with outbreaks occurring every 2–3 years. In developed countries, 5 out of 1,000 patients with VZ are hospitalized, and 2–3 cases of the disease per 100,000 infected people are fatal [2]. In the pre-vaccination era, VZ was a ubiquitous childhood infection in temperate countries. For example, in the United States, about 98% of the population was seropositive for VZV by the age of 20 years.
The first live attenuated vaccine (vOka) against this pathogen was obtained in 1974 through a series of passages of a wild-type clinical isolate of VZV (Oka). Later, this vaccine was improved, resulting in the release of Varivax (Merck Sharp & Dohme Corp.) [3]. The live attenuated vaccine consists of a mixture of different VZV genetic variants containing 42 single nucleotide polymorphisms that distinguish vOka from the parental wild-type Oka strain [2]. Due to the developed vaccine and the introduction of mandatory vaccination programs against VZV in different countries, hospitalization and mortality caused by VZ among children decreased by more than 90% [2, 3].
Varicella-zoster virus genome
Like all herpesviruses, VZV is a double-stranded DNA virus and has a 125,000-bp genome containing 72 open reading frames (ORFs) comprising 71 genes. Since the 3 genes are copies of other genes, the genome contains 68 unique genes. The viral genome consists of two main coding regions: unique long (UL) and unique short (US) ones. The UL region is flanked by short (≈ 88 bp) inverted repeats TRL and IRL, whereas the US region is flanked by long (7319 bp) TRS and IRS repeats. Five regions of the genome contain tandem direct repeats (R1, R2, R3, R4, and R5), which are short sequences with high G + C content [4, 5]. Three of these repeated sequences (R1, R2, and R3) are located in the coding region of the ORF11, ORF14, and ORF22 genes, respectively, and thus may influence protein functions. Two copies of R4 are located within the IR and TR repeats adjacent to the replication origin point (OriS), and R5 is located between ORF60 and ORF61 [5].
VZV genotyping
Until 2008, there was no uniform nomenclature for VZV typing. Early classification of VZV was based on the results of restriction fragment length polymorphism (RFLP) assay. The RFLP markers involved in these studies included the ORF38 (PstI), ORF54 (BglI), and ORF62 (SmaI) polymorphisms [6, 7]. Thus, most wild-type strains from the United States and Europe were characterized as PstI+BglI−; strains distributed in Asia and Africa – as BglI+; Oka-like wild-type strains from Japan – as PstI+/PstI–BglI+SmaI–; vaccine strains Oka – as PstI–BglI+SmaI+ [7, 8].
Attempts were made to identify VZV variants using complete genome screening for single nucleotide substitutions by heteroduplex mobility. This approach was used to evaluate substitutions in ORF1, ORF21, ORF50, and ORF54, which allowed identifying 4 major clades (genetic variants), named A, B, C, and J. Strains from Africa and Asia clustered in clade A, whereas clades B and C were predominantly composed of European strains. Subsequently, clade J was added to this genotyping scheme to account for Japanese strains [9, 10].
Another approach to genotyping is based on the single nucleotide polymorphisms assay in the gene sequences of 5 glycoproteins (gH, gI, gL, gB, gE), as well as the main transactivator gene IE62. It allowed classifying VZV strains into 4 clades: A, B, C, and D. Clades A and D were represented by isolates selected in North America and Europe, clades B and C consisted of VZV representatives common in Singapore and Japan, respectively [11, 12]. It should be noted that although the nomenclature of the clades from this study is much similar to the nomenclature used in the studies by W. Barrett-Muir et al. [8, 9], it has no correlation between the considered typing methods.
More recent phylogenetic analyses including both complete VZV genomes and their fragments have allowed the attribution of genomic variations to specific genetic variants and putatively recombinant viruses. Applying multilocus analysis of polymorphisms in the genomic sequences of VZV, 3 main genetic variants (clades) were identified: E (European), J (Japanese), and M (mosaic) [13]. Subsequently, the M genetic variant was divided into 4 separate variants: M1, M2, M3, and M4 [13, 14]. Strains belonging to genetic variants M1 and M2 were most common in tropical regions, genetic variant E — in temperate latitudes, and genetic variant J — in Japan. In some studies, a strain with the M3 genetic variant was found in the USA, and strains with the M4 genetic variant were found in Spain and France [14, 15].
Since the nomenclature of VZV clades/genetic variants was based on various molecular typing methods, a new universal nomenclature was introduced in 2008, dividing the genetic variants into 5 major clades (1–5) with 2 conditional (candidate) clades (VI and VII) [16] Also, now, in order to isolate VZV into a separate clade, the latter is required to include at least 2 representatives for which complete genome sequences have been obtained. Otherwise, such a clade is considered a candidate and is designated by Roman numerals.
By sequencing whole genomes from clinical VZV specimens collected in Germany, strains belonging to clades 1, 3, and 5, as well as strains that did not belong to any known clade, were discovered. Therefore, these representatives (1483/2005 and 457/2008) were allocated to separate candidate clades – VIII and IX, respectively [17]. Later, during the investigation of recombination events between the representatives of different VZV clades, a strain was found that grouped together with the only representative of clade VI in the phylogenetic tree. Thus, in 2015, candidate clade VI was converted to major clade 6 [18]. In 2017, the previously used VZV genotyping scheme was improved, which allowed identifying new representatives of candidate clade IX (one complete and one partial genome). However, only one isolate belonging to this clade (JN704710) was previously identified and described. The availability of the complete genome sequence of isolate KY037798 identified in this study met the requirement for conversion of candidate clade IX to major clade 9 [19].
As of today, VZV diversity is represented by 8 clades: 7 major clades (1–6 and 9) and 1 candidate clade VIII; clade VII has become irrelevant due to the lack of new isolates. The major VZV clades tend to be geographically confined to specific regions. In particular, clades 1, 3, and 6 are mainly spread in Europe, North America, and Australia, clade 2 – mainly in Asia. Clades 4 and 5 tend to be spread in multiple regions around the world; clade 5 is the only one reported to circulate in Africa [20]. Despite the vast amount of information on VZV clades circulating around the world, the distribution of VZV clades in Russia is not sufficiently clarified. There is virtually no relevant information on this subject in the scientific literature. In China's northern Xinjiang region, which borders with Russia, clades 1 and 3 are reported to be dominant [21].
As the first step towards obtaining reliable data on representatives of various VZV clades circulating in Russia, we have developed a specific primer panel that allows VZV typing in accordance with the scheme proposed by N.J. Jensen et al. [19]. The panel includes 6 pairs of primers covering short (about 200 bp) regions of open reading frames: ORF21, ORF22, ORF 29, ORF38, ORF55, ORF67, which contain 7 single nucleotide substitutions, the unique compositions of which make it possible to determine whether the virus belongs to one of the known clades. Thanks to the modification of primers by fusing them with Nextera adapter sequences along with the ability to sequence the resulting DNA libraries on the Illumina platform with reagent kits for short reads, it is possible to obtain typing results within 48 hours. Thus, the developed primer panel allows reducing financial and time costs while simultaneously increasing the number of specimens in one run of the device for high-throughput sequencing.
The solution that we developed, designed for rapid screening of a large number of clinical specimens, was tested on specimens from Moscow, Moscow Region, and Stavropol Territory, which made it possible to consider the representation of VZV in individual regions of the Russian Federation as a first approximation.
Materials and Methods
VZV specimens
The study used 75 VZV specimens collected from June 2022 to July 2023. Clinical specimens of viruses were obtained from patients mainly from Moscow (61 specimens), as well as from the Moscow Region (9 specimens) and Stavropol Territory (5 specimens). Biological material was collected after obtaining the voluntary consent of patients or their representatives. The study was approved at a meeting of the Local Ethics Committee of the Central Research Institute of Epidemiology of Rospotrebnadzor on May 24, 2022 (Minutes No. 124).
Isolation of DNA and obtaining fragments of VZV genomes
The viral DNA was extracted from clinical specimens using a Ribo-PREP reagent kit (AmpliSens). The resulting DNA was then used as a template for PCR in order to amplify fragments of the VZV genome. Regions of open reading frames (ORF21, ORF22, ORF 29, ORF38, ORF55, ORF67) containing discriminating mutations, according to which it is possible to perform VZV typing, were obtained using a developed set of primers (Table 1) mixed into a pool (Table 2).
Table 1. List of primers used to amplify VZV genome fragments
Primer | Primer sequence with Nextera adapter sequence (5'→3') |
VZV_1_fwd | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGgcggttttaacttcacaatgtaat |
VZV_1_rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGctcatctggacgaagcca |
VZV_2.2_fwd | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGttacccacaagcacgtcag |
VZV_2.2_rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGtcatcaaaccatgttaaccctc |
VZV_3_fwd | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGaatatgttacggggacctttga |
VZV_3_rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGcaaacaccccaataggttga |
VZV_4_fwd | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGgccatataccgcaacaactg |
VZV_4_rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGcctcgccataaagccactac |
VZV_5_fwd | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGccaccacggtggactatg |
VZV_5_rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGgaggagaccgtacgcga |
VZV_6_fwd | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGctttgatcttcaagggcgac |
VZV_6_rev | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGcgctatagtttgtcccggt |
Note. Capital letters indicate Nextera adapter sequences fused to primers (indicated in lower case letters).
Table 2. Composition of the mixture of primers for VZV typing
Primer | Primer concentration in mix, pmol/μl | Concentration after 2-fold dilution, pmol/μL |
VZV_1_fwd | 10,7 | 5,35 |
VZV_1_rev | 10,7 | 5,35 |
VZV_2.2_fwd | 7,1 | 3,55 |
VZV_2.2_rev | 7,1 | 3,55 |
VZV_3_fwd | 10,7 | 5,35 |
VZV_3_rev | 10,7 | 5,35 |
VZV_4_fwd | 9,5 | 4,75 |
VZV_4_rev | 9,5 | 4,75 |
VZV_5_fwd | 7,1 | 3,55 |
VZV_5_rev | 7,1 | 3,55 |
VZV_6_fwd | 4,7 | 2,35 |
VZV_6_rev | 4,7 | 2,35 |
Amplification was carried out in 25 µl of reaction mixture with the following composition: 10 µl PCR-mix-2 blue (AmpliSens), 1.8 µl of 4.4 mM deoxynucleoside triphosphate (AmpliSens), 1 µl of primer mixture, 5 µl of DNA template; the reaction mixture was brought to the required volume with H2O mQ. For amplification, the following time and temperature protocol was applied: 95ºC for 2 min; then 40 cycles: 95ºC for 15 s, 60°C for 30 s, 72ºC for 1 min; final elongation: 72ºC for 3 min. The target fragments of VZV genomes were identified by ethidium bromide fluorescence in 1.7% agarose gel. The results of electrophoretic analysis were visualized using GelDoc EZ (Bio-Rad).
Preparation of libraries for sequencing
The PCR product was purified from the reaction mixture using AMPureXP beads (Beckman Coulter) in a ratio of 1 : 0.8 (specimen : particles). The primers used to amplify VZV genome segments are modified by fusing them with Nextera adapters (Illumina), resulting in the amplicons being flanked by specific sequences that facilitate preparation of the libraries. Barcoding was performed using 10 μl of PCR-mix-2 blue (AmpliSens), 1 μl of EvaGreen (Biotium) as a fluorescent intercalating dye, 2 μl of each barcode (5 pmol) and 8 μl of purified PCR product. The following time and temperature protocol was used for barcoding: 98ºC for 30 s; 12 cycles: 98ºC for 10 s, 65ºC for 1 min 15 s. The DNA concentration after barcoding was measured on a Qubit 4.0 fluorimeter using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). The finished libraries were pooled and purified using AMPureXP magnetic beads (Beckman Coulter). The lengths of the finished libraries were identified using a 2100 Bioanalyzer automated electrophoresis system (Agilent Technologies).
Sequencing of VZV libraries
Sequencing was carried out on the Illumina MiSeq platform using the MiSeq Reagent Kit v2 (300 cycles) and MiSeq Reagent Kit v3 (600 cycles). At the same time, less than 0.5% of the MiSeq run was allocated to one specimen.
Determination of DNA sequences and genetic variants of specimens
After receiving “raw” data from the device, DNA reads were trimmed from the above-described primer sequences using the fastp 0.23.0 software tool [22]. Next, using bwa 0.7.17 [23], the reads were aligned (“mapped”) to the reference sequence of virus NC_001348.1, sorted and indexed using samtools 1.15.1 [24]. The resulting alignments were presented as draft genome assemblies using samtools 1.15.1 and an in-house script, and then remapped using MAFFT v7.490 [25] to obtain final assemblies (consensus sequences).
According to the above-mentioned nomenclature of viral clades [19], successful genotyping needs only 7 positions, the combination of variations (single-nucleotide substitutions) in which indicates a particular clade (Table 3). The primers that we constructed (Table 1) are designed to include these variations.
Table 3. The genotyping scheme proposed by N.J. Jensen et al. [19] (adapted)

Note. The least common “discriminatory” variants are highlighted in black.
Results
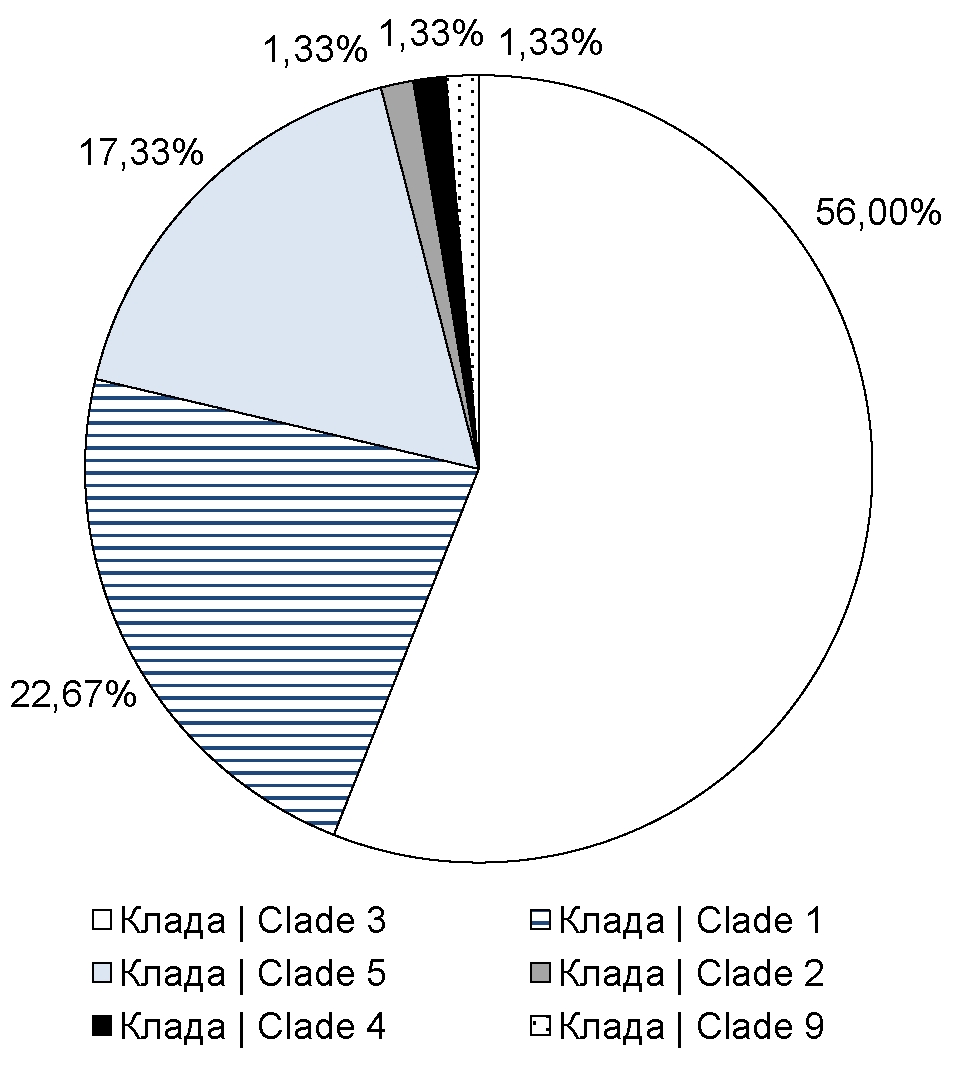
The primer panel that we developed, described in the previous section, covers short segments of 6 open reading frames: ORF21, ORF22, ORF29, ORF38, ORF55 and ORF67, which contain single-nucleotide substitutions that are targets for VZV typing. Using this panel, we sequenced clinical specimens of VZV obtained from patients from Moscow, Moscow Region, and Stavropol Territory. The resulting open reading frame segment sequences were sequenced on an Illumina MiSeq device with an average coverage of approximately 104. The minimum coverage for each segment was approximately 300 reads. Consensus sequences were obtained by processing the raw sequencing data using previously described publicly available bioinformatics tools and an in-house script. The assembled consensus sequences were used as input to the developed algorithm that performs VZV genotyping based on discriminatory mutations. Using this genotyping algorithm, we found that 17 specimens (22.67% of the total number of analyzed sequences) belonged to clade 1, 42 specimens (56%) belonged to clade 3 (Figure), 13 (17.33%) specimens belonged to clade 5. In addition, among all sequenced specimens, we identified single cases of infections caused by VZVs of clade 2 (1.4%), 4 (1.4%), and 9 (1.4%) (Figure).

Representation of various clades of VZV in the Russian Federation
Discussion
In this study, we developed and optimized a technique for rapid typing of a large number of VZV specimens. Moreover, using the described genotyping protocol, we obtained primary data on the VZV clades that have become widespread in Moscow, Moscow Region, and Stavropol Territory. Using next-generation sequencing technologies, we found that in the regions under consideration, viruses belonging to clades 1, 3, and 5 have become widespread. Also, we found single specimens belonging to clade 2, clade 4, and clade 9, which, compared to other clades, are a minor part of VZVs circulating in these regions of the Russian Federation. It is worth clarifying that the resulting picture of the distribution of clades is the most plausible for Moscow, since the largest number of clinical specimens were obtained from this region of the Russian Federation. In the future, if appropriate, it is planned to both increase the total number of specimens from the regions considered in this study and include other regions of Russia in the study.
At the moment, the data obtained as a result of our study only allow us to assume a largely similar picture of the distribution of VZV representatives in the studied regions of the Russian Federation and in European countries [20], and also correspond to the earlier reported data on circulating VZV clades in the region of China bordering Russia [21]. However, in the studied regions, apparently, a significantly larger proportion is occupied by viruses belonging to clade 3; in addition, the percentage of representation of clade 5 viruses in these regions of the Russian Federation is also higher than in European countries.
Conclusion
VZV, the etiologic agent of varicella and herpes zoster, is divided into several clades characterized by distinct genetic properties. We have successfully used next-generation sequencing technique to genotype VZV, which includes a custom primer panel and genotyping algorithm, an approach that can speed up the data collection process and reduce specimen preparation costs, especially when increasing the number of specimens per sequencing run. Using this methodology, we found that VZV clades 1, 3, and 5 predominated in Moscow, Moscow Region, and Stavropol Territory. The identification of these VZV clades provides valuable epidemiological data useful for shaping public health strategies and lifts the veil off the prevalence of VZV clades in some regions of the Russian Federation.
Note that this study is a “pilot” one and is mostly methodological in nature. It is also worth mentioning that in this study we demonstrated the prevalence of VZV clades only for 3 of the many regions of the Russian Federation, thus, the issue of the global representation of various virus clades in our country remains open, and therefore future research should be continued and expanded in order to form a reliable picture of the distribution of VZV clades in Russia.
About the authors
Maksim I. Nadtoka
Central Research Institute of Epidemiology
Author for correspondence.
Email: maximnadtoka@gmail.com
ORCID iD: 0009-0002-3217-0963
junior researcher, Genomics research laboratory, Central Research Institute for Epidemiology
Russian Federation, MoscowVladislav G. Lysenkov
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0002-1468-1631
bioinformatician, Genomics research laboratory, Central Research Institute for Epidemiology
Russian Federation, MoscowMatvei R. Agletdinov
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0003-2249-7196
bioinformatician, Genomics research laboratory, Central Research Institute for Epidemiology
Russian Federation, MoscowAndrey A. Mishkin
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0001-6911-6296
junior researcher, Central Research Institute for Epidemiology
Russian Federation, MoscowNatalia M. Afonina
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0002-3205-4025
Cand. Sci. (Med.), researcher, Laboratory for immunological prophylaxis, Central Research Institute for Epidemiology
Russian Federation, MoscowAntonina A. Ploskireva
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0002-3612-1889
D. Sci. (Med.), Deputy director for clinical work, Central Research Institute for Epidemiology
Russian Federation, MoscowIrina V. Mikheeva
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0001-8736-4007
D. Sci. (Med.), Professor, Head, Laboratory for immunological prophylaxis, Central Research Institute for Epidemiology
Russian Federation, MoscowKamil F. Khafizov
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0001-5524-0296
PhD, Head, Genomics research laboratory, Central Research Institute for Epidemiology
Russian Federation, MoscowVasily G. Akimkin
Central Research Institute of Epidemiology
Email: maximnadtoka@gmail.com
ORCID iD: 0000-0003-4228-9044
D. Sci. (Med.), Professor, Academician of the Russian Academy of Sciences, Director,Central Research Institute for Epidemiology
Russian Federation, MoscowReferences
- Laing K.J., Ouwendijk W.J.D., Koelle D.M., Verjans G.M.G.M. Immunobiology of varicella-zoster virus infection. J. Infect. Dis. 2018;218(Suppl. 2):S68–74. DOI: https://doi.org/10.1093/infdis/jiy403
- Gershon A.A., Breuer J., Cohen J.I., et al. Varicella zoster virus infection. Nat. Rev. Dis. Primers. 2015;1:15016. DOI: https://doi.org/10.1038/nrdp.2015.16
- Schmid D.S., Jumaan A.O. Impact of varicella vaccine on varicella-zoster virus dynamics. Clin. Microbiol. Rev. 2010; 23(1):202–17. DOI: https://doi.org/10.1128/cmr.00031-09
- Schmidt-Chanasit J., Sauerbrei A. Evolution and world-wide distribution of varicella-zoster virus clades. Infect. Genet. Evol. 2011;11(1):1–10. DOI: https://doi.org/10.1016/j.meegid.2010.08.014
- Depledge D.P., Sadaoka T., Ouwendijk W.J.D. Molecular aspects of varicella-zoster virus latency. Viruses. 2018;10(7):349. DOI: https://doi.org/10.3390/v10070349
- LaRussa P., Lungu O., Hardy I., et al. Restriction fragment length polymorphism of polymerase chain reaction products from vaccine and wild-type varicella-zoster virus isolates. J. Virol. 1992;66(2):1016–20. DOI: https://doi.org/10.1128/jvi.66.2.1016-1020.1992
- Loparev V.N., Argaw T., Krause P.R., et al. Improved identification and differentiation of varicella-zoster virus (VZV) wild-type strains and an attenuated varicella vaccine strain using a VZV open reading frame 62-based PCR. J. Clin. Microbiol. 2000;38(9):3156–60. DOI: https://doi.org/10.1128/jcm.38.9.3156-3160.2000
- Quinlivan M., Hawrami K., Barrett-Muir W., et al. The molecular epidemiology of varicella-zoster virus: evidence for geographic segregation. J. Infect. Dis. 2002;186(7):888–94. DOI: https://doi.org/10.1086/344228
- Barrett-Muir W., Scott F.T., Aaby P., et al. Genetic variation of varicella-zoster virus: evidence for geographical separation of strains. J. Med. Virol. 2003;70(Suppl. 1):S42–7. DOI: https://doi.org/10.1002/jmv.10319
- Muir W.B., Nichols R., Breuer J. Phylogenetic analysis of varicella-zoster virus: evidence of intercontinental spread of genotypes and recombination. J. Virol. 2002;76(4):1971–9. DOI: https://doi.org/10.1128/jvi.76.4.1971-1979.2002
- Faga B., Maury W., Bruckner D.A., Grose C. Identification and mapping of single nucleotide polymorphisms in the varicella-zoster virus genome. Virology. 2001;280(1):1–6. DOI: https://doi.org/10.1006/viro.2000.0775
- Wagenaar T.R., Chow V.T., Buranathai C., et al. The out of Africa model of varicella-zoster virus evolution: single nucleotide polymorphisms and private alleles distinguish Asian clades from European/North American clades. Vaccine. 2003;21(11-12):1072–81. DOI: https://doi.org/10.1016/s0264-410x(02)00559-5
- Loparev V.N., Gonzalez A., Deleon-Carnes M., et al. Global identification of three major genotypes of varicella-zoster virus: longitudinal clustering and strategies for genotyping. J. Virol. 2004;78(15):8349–58. DOI: https://doi.org/10.1128/jvi.78.15.8349-8358.2004
- Loparev V., Martro E., Rubtcova E., et al. Toward universal varicella-zoster virus (VZV) genotyping: diversity of VZV strains from France and Spain. J. Clin. Microbiol. 2007;45(2):559–63. DOI: https://doi.org/10.1128/jcm.01738-06
- Sergeev N., Rubtcova E., Chizikov V., et al. New mosaic subgenotype of varicella-zoster virus in the USA: VZV detection and genotyping by oligonucleotide-microarray. J. Virol. Methods. 2006;136(1-2):8–16. DOI: https://doi.org/10.1016/j.jviromet.2006.03.021
- Breuer J., Grose C., Norberg P., et al. A proposal for a common nomenclature for viral clades that form the species varicella-zoster virus: summary of VZV Nomenclature Meeting 2008, Barts and the London School of Medicine and Dentistry, 24-25 July 2008. J. Gen. Virol. 2010;91(Pt. 4):821–8. DOI: https://doi.org/10.1099/vir.0.017814-0
- Zell R., Taudien S., Pfaff F., et al. Sequencing of 21 varicella-zoster virus genomes reveals two novel genotypes and evidence of recombination. J. Virol. 2012;86(3):1608–22. DOI: https://doi.org/10.1128/jvi.06233-11
- Norberg P., Depledge D.P., Kundu S., et al. Recombination of globally circulating varicella-zoster virus. J. Virol. 2015; 89(14):7133–46. DOI: https://doi.org/10.1128/jvi.00437-15
- Jensen N.J., Rivailler P., Tseng H.F., et al. Revisiting the genotyping scheme for varicella-zoster viruses based on whole-genome comparisons. J. Gen. Virol. 2017;98(6):1434–8. DOI: https://doi.org/10.1099/jgv.0.000772
- Pontremoli C., Forni D., Clerici M., et al. Possible European origin of circulating varicella zoster virus strains. J. Infect. Dis. 2020;221(8):1286–94. DOI: https://doi.org/10.1093/infdis/jiz227
- Xu S., Chen M., Zheng H., et al. Nationwide distribution of varicella-zoster virus clades in China. BMC Infect. Dis. 2016; 16(1):542. DOI: https://doi.org/10.1186/s12879-016-1863-x
- Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. DOI: https://doi.org/10.1093/bioinformatics/bty560
- Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14): 1754–60. DOI: https://doi.org/10.1093/bioinformatics/btp324
- Li H., Handsaker B., Wysoker A., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16): 2078–9. DOI: https://doi.org/10.1093/bioinformatics/btp352
- Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic. Acids Res. 2002;30(14):3059–66. DOI: https://doi.org/10.1093/nar/gkf436
Supplementary files

