


Том 100, № 5 (2023)

- Год: 2023
- Выпуск опубликован: 22.11.2023
- Статей: 12
- URL: https://microbiol.crie.ru/jour/issue/view/180

ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
Изучение генетического разнообразия вируса ветряной оспы в отдельных регионах Российской Федерации при помощи высокопроизводительного секвенирования
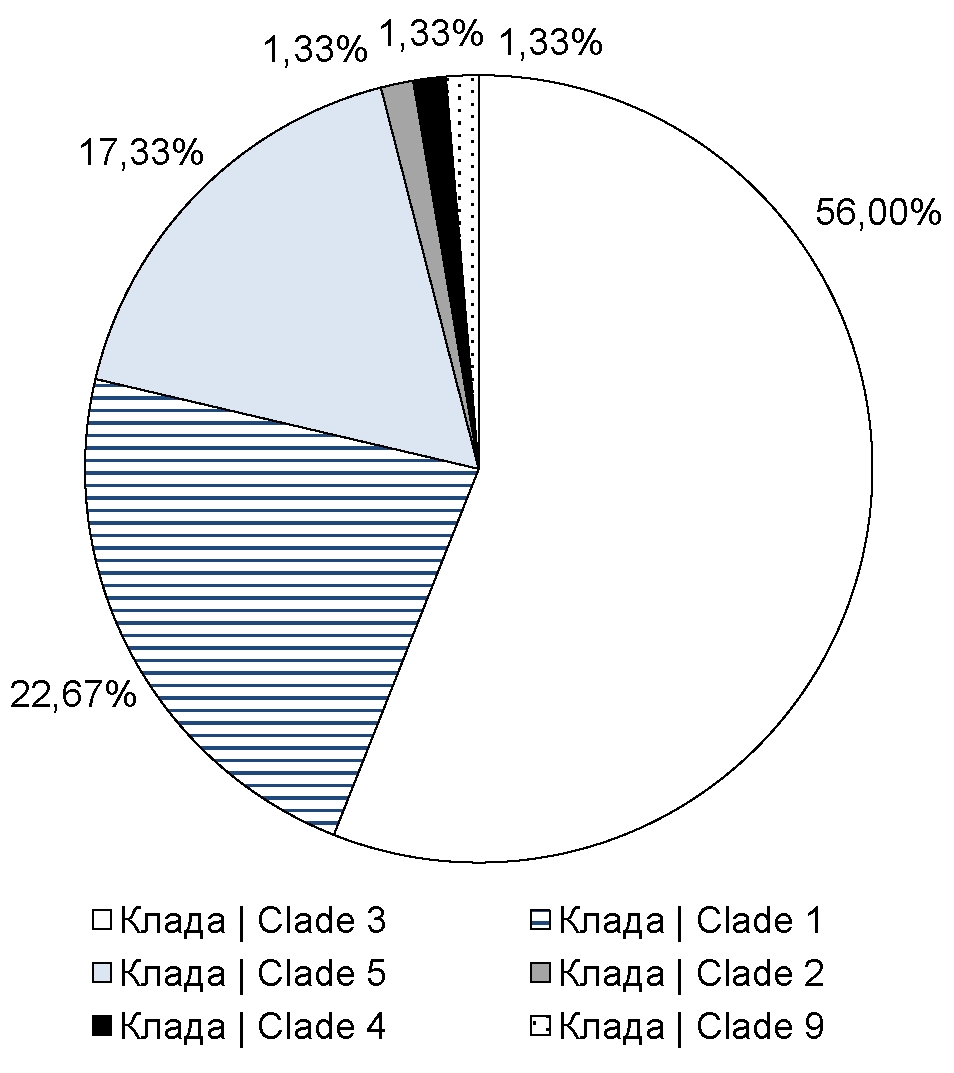
Аннотация
Введение. Вирус ветряной оспы (VZV) — возбудитель одноимённого заболевания и опоясывающего лишая, филогенетически подразделяется на 8 клад, для распространения которых характерна географическая привязка к тем или иным регионам мира. Для большинства стран установлены циркулирующие на их территориях клады VZV, однако для России аналогичная информация практически отсутствует.
Цель исследования — разработка эффективной методики типирования VZV с использованием технологий высокопроизводительного секвенирования для выявления распространённости различных клад VZV в Москве, Московской области и Ставропольском крае.
Материалы и методы. Для генотипирования VZV достаточно задействовать 7 нуклеотидных позиций, по уникальным сочетаниям которых возможно отнести вирус к одной из клад. Короткие участки нуклеотидных последовательностей открытых рамок считывания получали при помощи разработанного набора праймеров.
Результаты. Разработана и оптимизирована методика генотипирования VZV. При помощи данной методики получены первичные данные о распределении клад VZV в исследуемых регионах. Таким образом, было установлено, что в Москве и ряде других регионов распространены преимущественно 1, 3 и 5-я клады VZV.
Заключение. Разработанная методика, включающая праймерную панель и алгоритм генотипирования, позволяет произвести типирование VZV в короткие сроки при снижении затрат на пробоподготовку и одновременном увеличении количества образцов в одном цикле секвенирования. Результаты, полученные с использованием данного протокола, позволяют сделать предположение о том, что в Москве, Московской области и Ставропольском крае наибольшую представленность имеют клады 1, 3 и 5 VZV. Для подтверждения данной гипотезы требуется включить в последующие исследования большее количество клинических образцов, в том числе из других регионов страны.
 267-275
267-275



Анализ уровня продукции факторов инвазии InlA и InlB у изолятов Listeria monocytogenes, выделенных на территории Российской Федерации
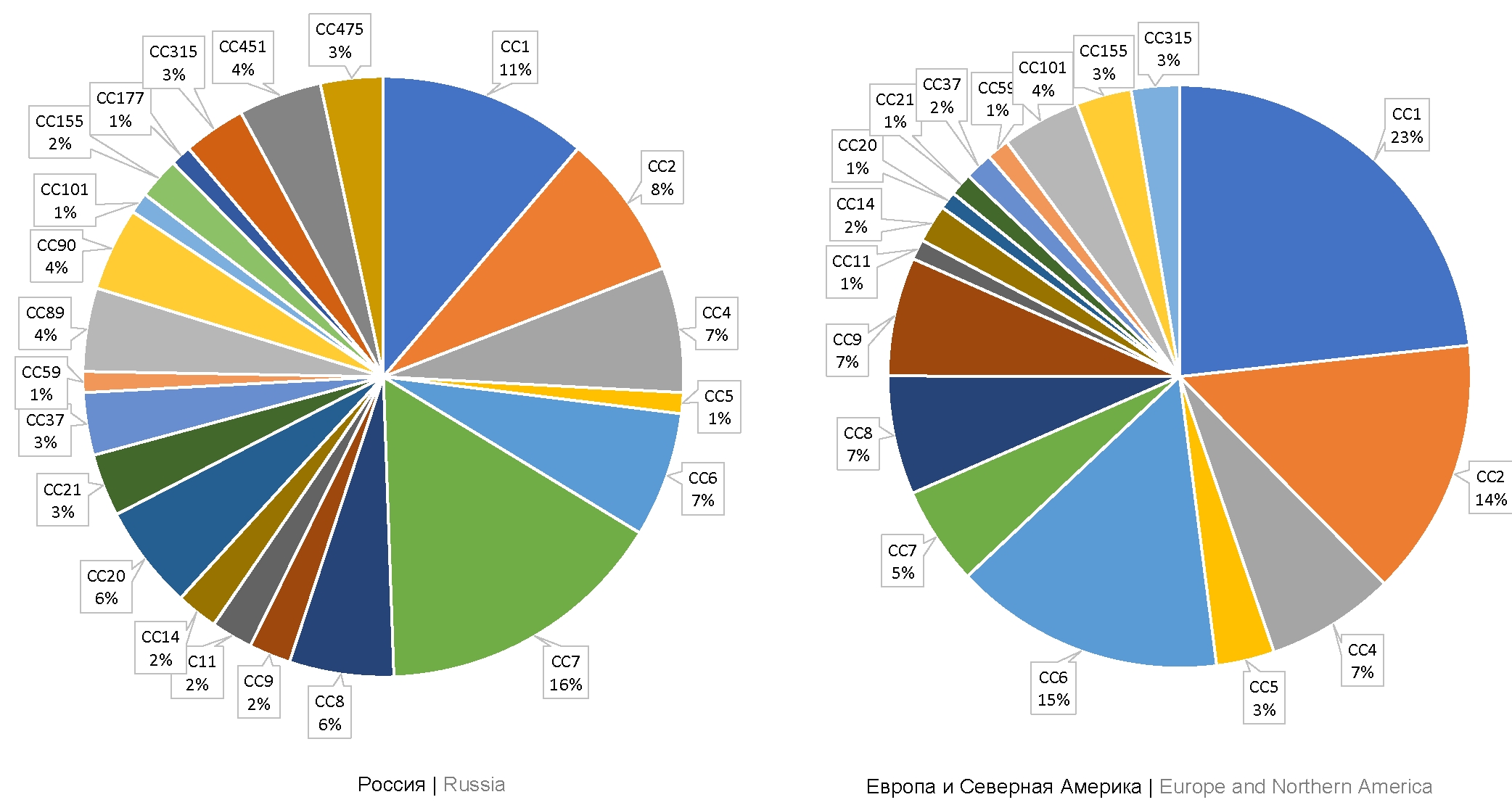
Аннотация
Актуальность. Listeria monocytogenes характеризуется наличием эпидемически высоковирулентных клонов. Инвазия в непрофессиональные фагоциты — ключевой момент листериозной инфекции. Формирование высоковирулентных клонов обусловлено повышенной продукцией и/или наличием определённых изоформ факторов инвазии белков InlA и InlB.
Цель исследования — создать тест-систему для обнаружения InlA и InlB и на её основе оценить уровни продукции InlA и InlB у изолятов L. monocytogenes, относящихся к клональным группам с различным вирулентным потенциалом.
Материалы и методы. В работе использованы 32 штамма L. monocytogenes, относящихся к эпидемическим клонам ECII, ECIV, ECVII (клональные комплексы СС1, СС2, СС7) и гиповирулентному клональному комплексу СС9. Проведено секвенирование генов inlA и inlB. Для анализа уровня продукции белков InlA и InlB использован непрямой иммуноферментный анализ.
Результаты. Выявлена вариабельность InlA среди штаммов, относящихся к одному клональному комплексу: в том числе среди штаммов, принадлежащих к СС7, выявлены 3 изоформы InlA; из 8 штаммов, принадлежащих к СС9, у одного выявили стоп-кодон в гене inlA, приводящий к утрате функциональности белка InlA. Различия между аллелями inlB коррелировали с принадлежностью штаммов к конкретному клональному комплексу. Установлены различия в уровне продукции факторов инвазии. У штаммов, относящихся к СС9, уровень продукции InlA был в 2,5 раза ниже по сравнению со штаммами, относящимися к СС1, СС2 и СС7. Уровень продукции InlB был в среднем в 4 раза выше у штаммов, принадлежащих к филогенетически родственным СС1 и СС2, по сравнению со штаммами, относящимися к СС7 и СС9.
Заключение. Полученные результаты свидетельствуют о вариабельности основных факторов инвазии как между клональными комплексами, так и между штаммами одного комплекса. Повышенная продукция факторов инвазии InlA и InlB коррелирует с потенциальной вирулентностью штаммов.
276-286
Характеристика антибиотикорезистентности нетифоидных сальмонелл, циркулирующих на территории Российской Федерации в период с 2019 по 2022 год

Аннотация
Введение. Нетифоидные сальмонеллы вносят значительный вклад в заболеваемость кишечными инфекциями и характеризуются возрастанием доли штаммов, резистентных к антимикробным препаратам (АМП), в том числе к современным препаратам выбора (цефалоспорины III и фторхинолоны).
Цель работы — оценка фенотипической резистентности сальмонелл к различным классам АМП и определение связи между фенотипической резистентностью, серотипом, источником изоляции и характером заболеваемости.
Материалы и методы. Исследованы 752 неповторяющихся штамма сальмонелл из 2494 штаммов, выделенных из различных источников (клинический материал, пищевые продукты, окружающая среда), поступивших из 59 регионов России в период с 2019 по 2022 г. Фенотипическая резистентность к 22 антибиотикам из 11 CLSI-классов АМП оценена методом серийных разведений в бульоне (минимальная подавляющая концентрация). Проведено сравнение разнообразия профилей резистентности серотипов сальмонелл с использованием индекса Шеннона.
Результаты. Доминирующее положение по частоте изоляции занимают серотипы Salmonella Еnteritidis, S. Infantis, S. Muenchen, S. Typhimurium, S. Bovismorbificans, на которые приходилось 64,4% исследованных штаммов. Устойчивость по меньшей мере к одному из тестируемых антибиотиков проявляли 543 (72,2%) штамма, множественной лекарственной устойчивостью характеризовались 193 (25,7%) штамма. Резистентность к классам АМП характеризовалась следующим распределением: хинолоны (61,3%), тетрациклины (28,1%), пенициллины (19,1%), β-лактамные комбинированные препараты (18,6%), антагонисты фолатного пути (16,5%), фениколы (10,1%), аминогликозиды (5,6%), цефемы (4,7%), монобактамы (4,4%), липопептиды (3,9%). Резистентных штаммов к пенемам не выявлено. Показаны особенности резистентности сальмонелл по классам АМП в зависимости от источников выделения, серотипа сальмонелл и характера заболеваемости (групповая и спорадическая).
Выводы. Мониторинг фенотипической антибиотикорезистентности является важным инструментом эпидемиологического надзора в целях профилактики распространения резистентности бактерий к АМП.
287-301
Сравнительная оценка эффективности воздействия дезинфицирующих веществ на микроорганизмы в биоплёнке
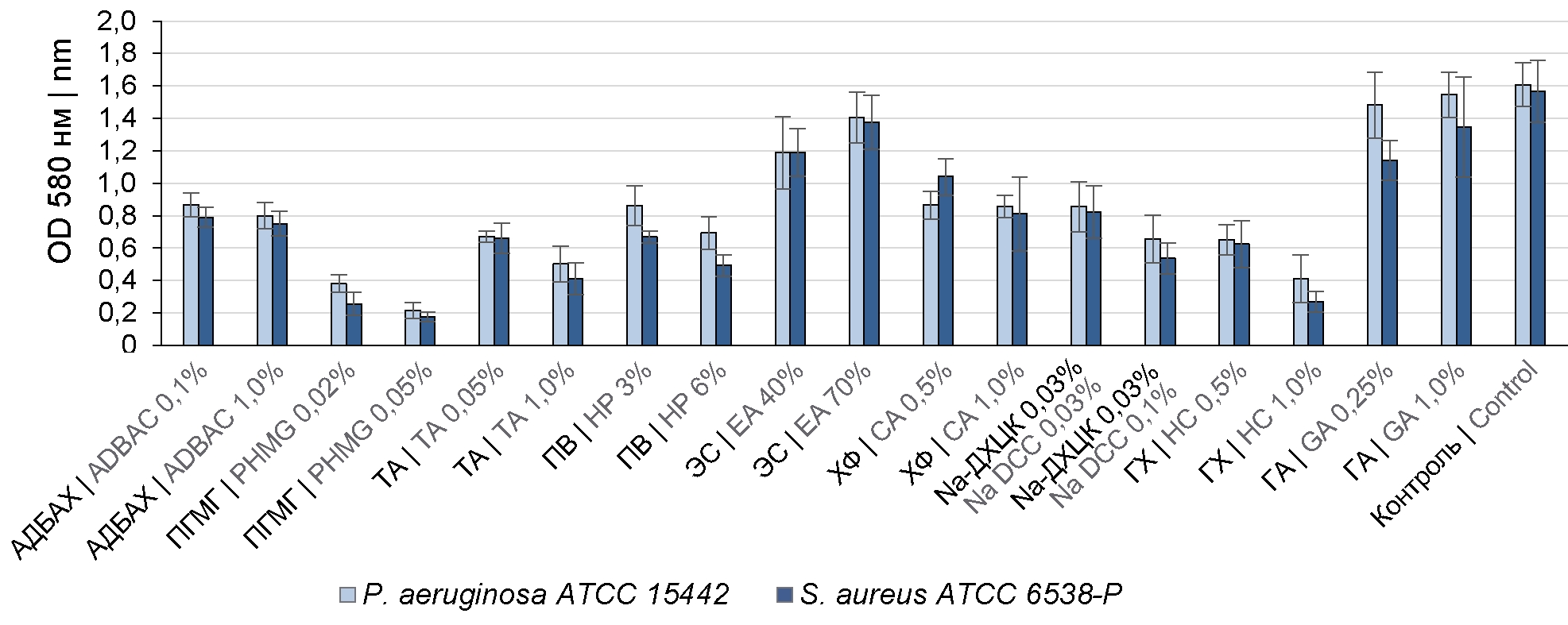
Аннотация
Введение. Бактерии в биоплёнке (БП) обладают повышенной устойчивостью к антибактериальным агентам, в том числе дезинфицирующим веществам (ДВ), однако степень эффекта варьирует в зависимости от приложенного воздействия. В связи с этим оценка эффективности основных ДВ в отношении микроорганизмов в БП представляет научный и практический интерес.
Целью исследования было изучение воздействия ДВ различных химических групп на грамположительные и грамотрицательные бактерии в составе БП.
Материалы и методы. Изучено действие ДВ: алкилдиметилбензиламмония хлорида (АДБАХ), третичного амина (ТА), полигексаметиленгуанидина хлорида (ПГМГ), перекиси водорода (ПВ), хлорамина (ХА), натриевой соли дихлоризоциануровой кислоты (Na-ДХЦК), гипохлорита натрия (ГХ), спирта этилового (ЭС), глутарового альдегида (ГА)) в отношении Pseudomonas aeruginosa ATCC 15442 и Staphylococcus aureus ATCC 6538-P в БП. БП культивировали в 96-луночных планшетах при 37оС в течение 24 ч, затем воздействовали на них растворами биоцидов. Эффективность воздействия ДВ оценивали на основании регистрации оставшихся жизнеспособных клеток и относительной плотности БП.
Результаты. Изученные штаммы бактерий образовывали умеренную БП, среднее количество жизнеспособных клеток в БП составило 6,51 ± 0,19 lg. Количество жизнеспособных клеток бактерий в составе БП снижалось на 4 lg и более под действием растворов ПВ в концентрации 6%, раствора Na-ДХЦК — 0,1% (по активному хлору), ГХ — 1% (по активному хлору), ХА — 1% (по препарату), ПГМГ — 0,05%, ТА — 1,0 %. При этом плотность БП снижалась на 70% и более. Растворы АДБАХ в концентрациях 0,1–1,0%, ТА — 0,05%, ПВ — 3%, раствор Na-ДХЦК — 0,05% (по активному хлору) обеспечивали снижение жизнеспособных клеток в БП на 2 lg. Эффективность воздействия хлорактивных соединений и ПВ повышалась при добавлении 0,5% сульфонола. Растворы ГА (0,25–1,00%) и ЭС (40–70%) были неэффективны в отношении микроорганизмов в БП.
Заключение. Для борьбы с микробными плёнками перспективны ДВ из группы окислителей (хлорактивные и кислородсодержащие), ТА и ПГМГ; применение АДБАХ как индивидуального соединения неэффективно; альдегиды и спирты для разрушения БП и уничтожения в ней микроорганизмов не пригодны.
302-309
Изоляция и генетический анализ вируса Чикунгунья из комаров Aedes aegypti и Aedes albopictus, отловленных в Центральной Америке
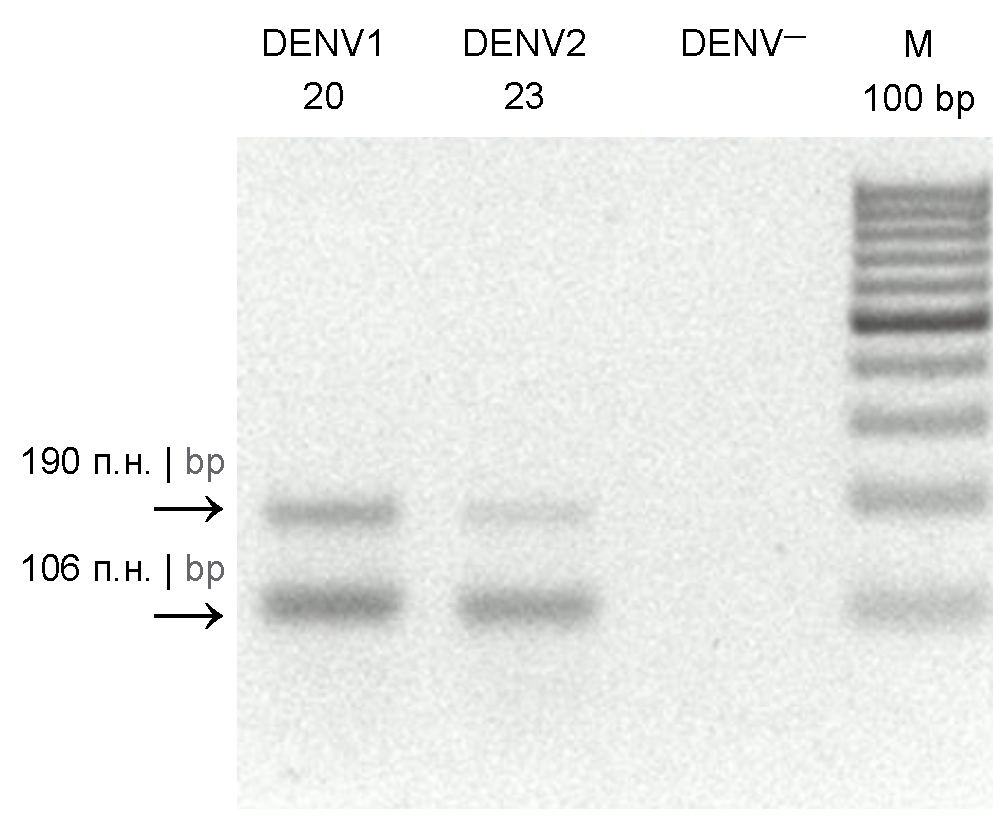
Аннотация
Введение. Ареал обитания комаров родов Aedes spp., Culex spp., Culiseta spp. распространяется на Южную и Центральную Америку, включая Никарагуа. Мониторинг за распространением комаров-переносчиков и оценка их инфицированности арбовирусами могут предоставить информацию о возможности появления новых или увеличении случаев уже регистрируемых заболеваний, изменении инфекционности вирусов для человека при смене переносчика возбудителя.
Целью настоящей работы были выделение и идентификация арбовирусов, принадлежащих к родам Flavivirus и Alphavirus, из комаров видов A. albopictus, A. aegypti, Culiseta spp., Culex spp., отловленных в лесах Никарагуа.
Материалы и методы. Комары A. albopictus, A. aegypti, Culiseta spp., Culex spp. были отловлены в 2021 г. в сухой сезон в лесной зоне в Никарагуа в четырех разных локациях. Комаров объединяли в пулы по 5–8 особей (всего 236 пулов). Методом полимеразной цепной реакции с обратной транскрипцией пулы анализировали на наличие вирусов Чикунгунья (ВЧ), денге, Зика и жёлтой лихорадки. Положительные пулы инокулировали в культуру клеток С6/36 с целью получения изолятов и их дальнейшего секвенирования.
Результаты. Вирус денге был выявлен только в комарах Aedes spp.: в 7 пулах — A. aegypti, в 1 — A. albopictus. ВЧ также был выявлен только в комарах Aedes spp.: в 3 пулах — A. aegypti, в 1 — A. albopictus. Секвенирование нуклеотидных последовательностей генов 6К, Е1, Е2 и NS1 ВЧ, выделенного из комаров A. albopictus, показало, что по сравнению с аналогичными последовательностями генов из изолятов ВЧ, выделенных из комаров A. aegypti, в области гена белка 6К обнаружено 4 нуклеотидных и столько же аминокислотных замен, в области Е1 — 16 нуклеотидных замен, 10 из которых приводили к аминокислотным заменам, в области Е2 — 14 нуклеотидных и 11 аминокислотных замен, в области NS1 — 33 нуклеотидные и 19 аминокислотных замен.
310-318
Сравнительный анализ уровня IgG в сыворотках крови больных COVID-19, вакцинированных «Гам-КОВИД-Вак», и здоровых доноров до пандемии
Аннотация
Введение. Измерение концентрации сывороточного иммуноглобулина G (IgG) используется в диагностике и мониторинге многих заболеваний. Поэтому важно изучить влияние пандемии COVID-19 на уровень IgG в популяции.
Целью работы был сравнительный анализ среднего уровня сывороточного IgG в образцах крови, полученных от госпитализированных пациентов с COVID-19 (n = 31), здоровых доноров до начала пандемии (n = 30) и доноров, вакцинированных препаратом «Спутник V» (не болевших COVID-19; n = 34).
Материалы и методы. IgG измеряли двумя методами твердофазного иммуноферментного анализа (ИФА): зарегистрированной российской системой «IgG общий-ИФА-БЕСТ» и сконструированным в лаборатории конкурентным ИФА, основанным на моноклональных биспецифических антителах к IgG человека и пероксидазе хрена.
Результаты и обсуждение. Отличий в среднем уровне сывороточного IgG в трёх группах не обнаружили (независимо от пола); оба метода дали соотносимые результаты. Однако система «IgG общий- ИФА-БЕСТ» выявила статистически значимые различия в концентрации сывороточного IgG в подгруппах пациентов-мужчин с разным уровнем антител к вирусному антигену к рецепторсвязывающему домену (receptor-binding domain, RBD): ниже и выше 400 BAU/мл. У 10 мужчин с RBD-антигеном < 400 BAU/мл содержание IgG было 14,3 ± 4,1 мг/мл, у 6 мужчин с RBD-антигеном > 400 BAU/мл — 6,9 ± 2,7 мг/мл.
Заключение. Средний уровень IgG в сыворотках крови, полученных до пандемии, не отличался от такового у здоровых доноров, вакцинированных препаратом «Спутник V», или пациентов, госпитализированных в связи с COVID-19. При этом обнаружено сравнительное снижение средней концентрации IgG у пациентов-мужчин с COVID-19 и уровнем анти-RBD антител > 400 BAU/мл. В учётом данных литературы об ассоциации снижения сывороточного IgG с тяжестью COVID-19 целесообразно сравнить выборки большего размера.
319-327
Мониторинг респираторных вирусных инфекций в 2011–2022 годах в Москве

Аннотация
Введение. Респираторные вирусы (РВ) циркулируют повсеместно и круглогодично. Долговременный мониторинг распространения возбудителей респираторных инфекций необходим для анализа соответствия диагностических систем современным вирусным изолятам, оценки рисков инфицирования и необходимости разработки и применения вакцин, а также для исследования взаимозависимости репродукции РВ при смешанных инфекциях.
Цель работы — исследование возбудителей острых респираторных вирусных инфекций (ОРВИ) в Москве в 2011–2022 гг. с помощью обратной транскрипции с последующей полимеразной цепной реакцией с гибридизационно-флуоресцентной детекцией продуктов в реальном времени (ОТ-ПЦР-РВ).
Материалы и методы. Мазки носоглотки 3908 пациентов с ОРВИ исследованы с помощью ОТ-ПЦР-РВ.
Результаты. Мониторинг распространения РВ в Москве показал циклические изменения частот с тремя доминирующими видами: вирусом гриппа А (до 31,3%), респираторно-синцитиальным вирусом (до 24,8%) и риновирусами человека (до 21,3%) в 2011–2020 гг., которые соответствовали данным из других регионов России и мировым тенденциям, и высокие частоты встречаемости SARS-CoV-2 (31,7%) в 2022 г. Рост доли неидентифицированных клинических образцов от 1,2 до 28,5% в 2022 г. свидетельствует о неполном соответствии диагностических систем современным изолятам РВ или о появлении новых видов или штаммов возбудителей. Зарегистрированы однонаправленные изменения динамики для 5 из 9 изучаемых РВ с коэффициентами корреляции 0,43–0,79. Высокие частоты смешанных ОРВИ (до 33,4%) наряду с неидентифицированными образцами не позволяют точно оценить риски инфицирования различными РВ в Москве, однако доказывают необходимость профилактики инфекционных заболеваний наиболее распространёнными РВ.
Заключение. Анализ динамики частот распространения РВ в Москве показал сохранение доминирующих видов: вируса гриппа А, респираторно-синцитиального вируса и риновирусов человека. В период вакцинации против COVID-19 увеличилась доля сезонных коронавирусов.
328-337
Молекулярно-эпидемиологический мониторинг популяции возбудителя туберкулёза в Архангельской области
Аннотация
Введение. На фоне улучшения основных эпидемиологических показателей (заболеваемость и смертность) по туберкулёзу (ТБ) в Архангельской области доля впервые выявленных больных ТБ с множественной лекарственной устойчивостью (МЛУ) возбудителя увеличилась с 18,7% в 2002 г. до 33,8% в 2018 г.
Целью исследования была генотипическая характеристика штаммов Mycobacterium tuberculosis, полученных от впервые выявленных больных ТБ в Архангельской области в 2018 г.
Материалы и методы. Изучены 89 штаммов M. tuberculosis, выделенных в 2018 г. от впервые выявленных больных ТБ. Принадлежность к генотипу Beijing, кластерам B0/W148 и Central-Asian/Russian Beijing определяли с помощью ПЦР-детекции специфических маркеров: инсерции IS6110 в области dnaA-dnaN, мутаций в кодонах 48 гена mutT4 (CGG>GGG) и 58 гена mutT2 (GGA>CGA), вставки IS6110 в области Rv2664-Rv2665 и Rv1359-Rv1360, замены G>A в гене sigE. Штаммы non-Beijing были сполиготипированы.
Результаты. Лекарственной устойчивостью обладали 41,6% (37/89), МЛУ — 33,7% штаммов. У 90% (27/30) МЛУ-штаммов устойчивость к рифампицину и изониазиду была обусловлена мутациями rpoB Ser531Leu и katG Ser315Thr. Выявлены генотипы M. tuberculosis: Beijing (67,4%), T (14,6%), Ural (4,5%), Haarlem (4,5%), LAM (2,3%) и CAS1-Delhi (1,1%). Среди штаммов Beijing преобладали кластеры Central-Asian/Russian (60%; 36/60) и B0/W148 (30%; 18/60). Большинство МЛУ-штаммов принадлежали к семейству Beijing (93,3%; 28/30), из которых 64,3% (18/28) и 21,4% (6/28) — к кластерам B0/W148 и Central-Asian/Russian соответственно.
Заключение. В гетерогенной популяции возбудителя ТБ Архангельской области наиболее распространёнными были штаммы генотипа Beijing, причём в 2018 г. его доля увеличилась до 67,4% (в 1998–1999 гг. — 40,4%). Среди МЛУ-штаммов доля Beijing достигла 93,3%, из них более половины (64,3%) принадлежали к эпидемиологически и клинически значимому в России кластеру B0/W148.
338-345
Анализ изменения генома геновариантов Vibrio cholerae О1 El Tor в современный период пандемии холеры
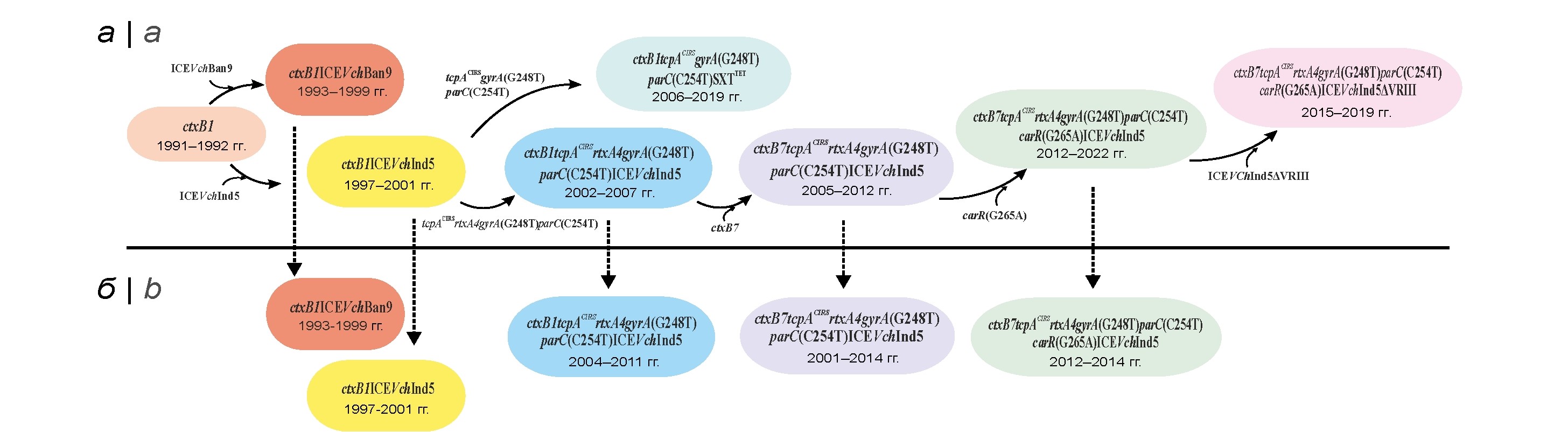
Аннотация
Введение. Вариабельность генома генетических вариантов возбудителя холеры El Tor обусловила появление штаммов, несущих мутации в различных генах патогенности и лекарственной устойчивости. Такая ситуация требует оценки направления этих измененений для прогнозирования патогенного потенциала ранее неизвестных вариантов и своевременной разработки новых средств диагностики и профилактики.
Цель работы — анализ динамики изменения генов патогенности и лекарственной устойчивости генетических вариантов Vibrio choleraе El Tor из эндемичных по холере стран и России.
Материалы и методы. Использовали секвенированные нуклеотидные последовательности полных геномов 104 штаммов V. cholerae El Tor, взятых из баз данных NCBI GenBank и European Nucleotide Archive, а также полногеномные сиквенсы, полученные нами. Анализ нуклеотидных последовательностей выполняли с помощью программы UGEN v.45.1. Для построения дендрограммы по алгоритму максимальной экономии применяли программный пакет «BioNumerics v. 7.6» на основе множественного выравнивания, полученного с помощью программы «Snippy 4.6.0».
Результаты. Cопоставлены секвенированные геномы 103 штаммов геновариантов, выделенных на территории 9 эндемичных стран Азии и Африки, а также в России в 1991–2022 гг. Показано, что процесс изменения генома геновариантов был многоступенчатым и происходил за счет последовательного накопления точечных мутаций в ключевых (ctxB и tcpA) и дополнительных (rtxA) генах патогенности и коровых генах резистентности к антибиотикам (gyrA, parC и carR), а также делецией в мобильном элементе SXT. Наиболее важным стало изменение в гене ctxB и появление новых геновариантов с аллелем ctxB7, вытеснивших ранее сформированные штаммы. Анализ измененных участков генома 83 штаммов геновариантов из эндемичных регионов выявил 8 генотипов, тогда как штаммы (21 изолят), завезённые в Россию, относились лишь к 5 генотипам, включая высоковирулентные геноварианты с аллелем ctxB7 и утраченным биоварспецифическим свойством PolR за счёт мутации гена carR. Установленная тесная филогенетическая связь геновариантов, выявленных в России, со штаммами из эндемичных стран Азии подтверждает их завоз из этого региона.
Заключение. Показано последовательное возникновение и накопление новых мутаций в генах патогенности и лекарственной устойчивости в геноме геновариантов в эндемичных регионах, что приводит к изменению их эпидемически важных свойств. Установлен завоз в Россию новых геновариантов с высокой вирулентностью, что указывает на необходимость постоянной оценки изменений генома этого патогена для своевременной разработки адекватных средств генодиагностики и профилактики.
346-357
Анализ циркуляции коксакивируса А6 в субъектах Дальневосточного федерального округа Российской Федерации в 2014–2019 годах
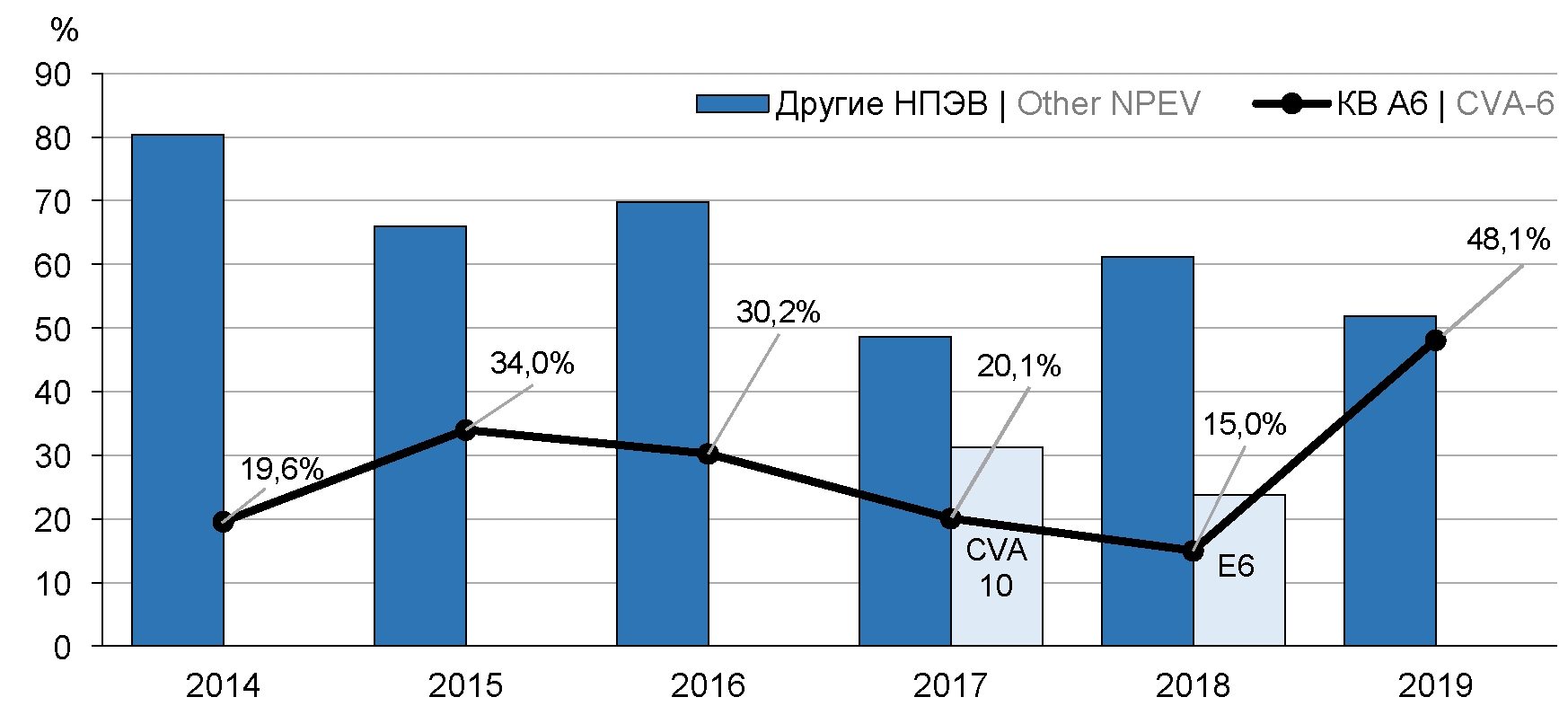
Аннотация
Введение. Молекулярно-эпидемиологический мониторинг за энтеровирусной инфекцией (ЭВИ) в субъектах Российской Федерации показал, что коксакивирус А6 (КВ-А6) на протяжении последних лет был одним из доминирующих типов энтеровирусов (ЭВ), циркулировавших среди населения страны, и в отдельные годы явился причиной большинства вспышек ЭВИ.
Цель работы — провести анализ циркуляции КВ-А6 в субъектах Дальневосточного федерального округа (ДФО) в 2014–2019 гг. с использованием молекулярно-генетических методов.
Материалы и методы. Биологический материал, поступавший из 9 субъектов ДФО, исследовали методом полимеразной цепной реакции с обратной транскрипцией для обнаружения РНК ЭВ. Проводили амплификацию положительных образцов для получения нуклеотидных последовательностей фрагментов генов VP1 и VP2 для дальнейшего установления типа ЭВ. Для молекулярно-генетического анализа дальневосточных штаммов КВ-А6 получены нуклеотидные последовательности фрагментов генов VP1 и 3Dpol. Построение филогенетических деревьев осуществляли с использованием Байесовых филогенетических методов.
Результаты. C 2014 по 2019 г. получены 1773 нуклеотидные последовательности неполиомиелитных ЭВ 43 типов, циркулировавших в ДФО, при этом основная часть сиквенсов принадлежала КВ-А6 (524; 29,5%). В годы наибольшей идентификации КВ-А6 в субъектах ДФО наблюдались подъёмы заболеваемости ЭВИ с регистрацией вспышечных очагов. Из клинических проявлений ЭВИ, вызванных КВ-А6, в ДФО преобладали герпангина и экзантемные формы. Филогенетический анализ показал принадлежность дальневосточных штаммов КВ-А6 к превалирующему во всём мире субгенотипу D3, а также циркуляцию в анализируемый период времени нескольких рекомбинантных форм КВ-А6 (RF-A, -H, -L, -N, -R).
Заключение. Установленное генетическое разнообразие штаммов КВ-А6, циркулировавших в субъектах ДФО в 2014–2019 гг., требует дальнейшего изучения для получения новых знаний о молекулярной эпидемиологии КВ-А6 и совершенствования системы эпидемиологического надзора за ЭВИ.
358-368
Филодинамическая характеристика гена LMP-1 вируса Эпштейна–Барр, изолированного на территории Нижегородской области
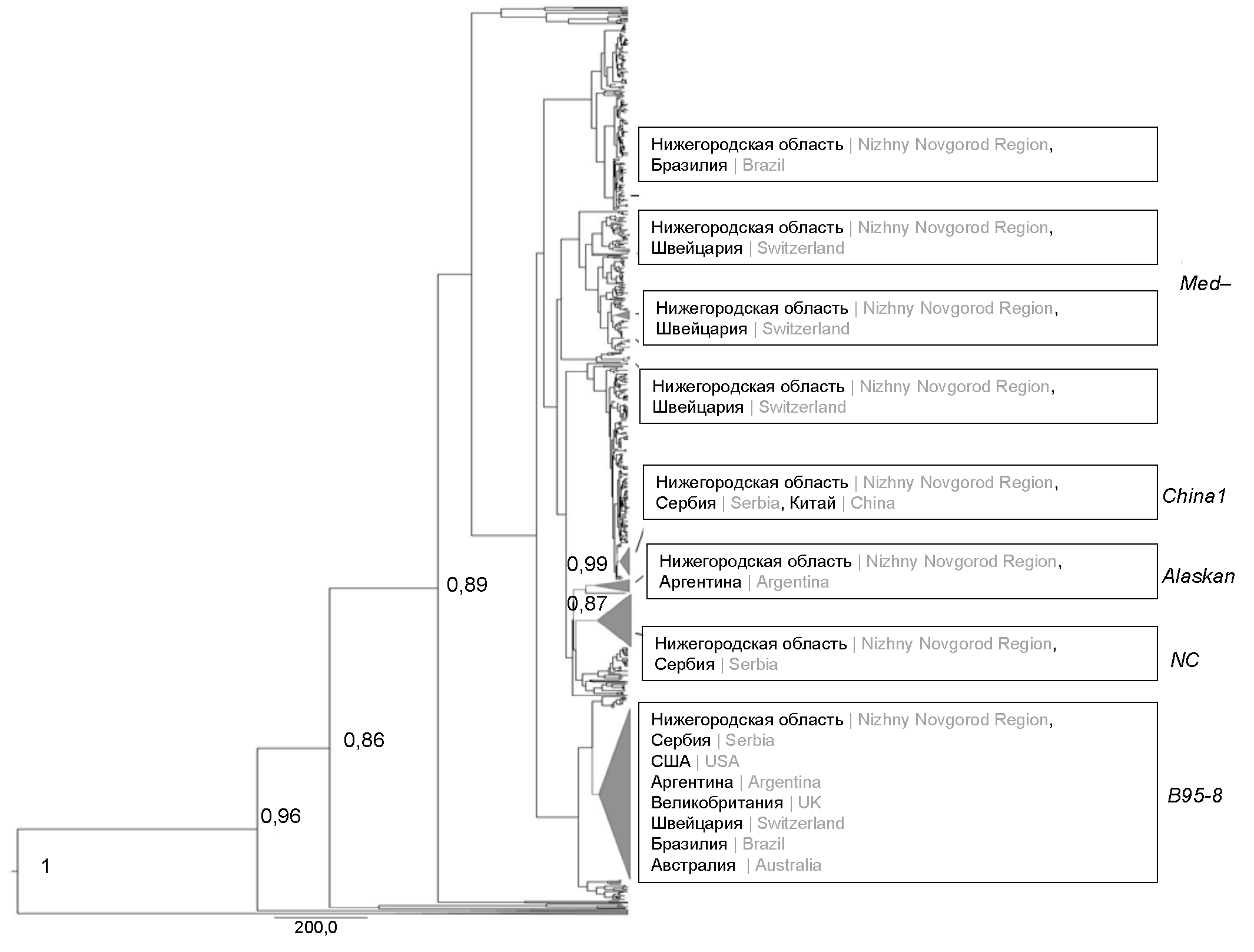
Аннотация
Введение. Вирус Эпштейна–Барр (ВЭБ) относится к числу самых распространённых герпесвирусов и обладает выраженным генетическим полиморфизмом. Изучение филодинамических характеристик вируса является важным аспектом исследования эволюционных изменений гена LMP-1 и их последствий.
Цель — филодинамический анализ нижегородских изолятов ВЭБ на основе C-концевого фрагмента гена LMP-1.
Материалы и методы. В исследование были включены 158 изолятов ВЭБ, полученных из лейкоцитов крови и слюны детей 1–17 лет с диагнозом «инфекционный мононуклеоз, вызванный ВЭБ» (n = 68) и условно здоровых детей сопоставимого пола и возраста (n = 29). Геноварианты LMP-1 были получены с помощью метода секвенирования по Сэнгеру. Сравнительный анализ аминокислотных последовательностей проводили в программе «MEGA X», филодинамический анализ полученных нуклеотидных последовательностей и изолятов, депонированных в GenBank, — в пакете программ «BEAST v. 1.10.4», рекомбинационный анализ — в программе «Simplot».
Результаты. Получены и депонированы в базу данных GenBank 158 нуклеотидных последовательностей С-концевого фрагмента гена LMP-1 нижегородских изолятов ВЭБ. Установлено время циркуляции ближайшего общего предка для модифицированных геновариантов B95-8 с мутациями G212S + E328Q + S366T и NC с заменой D250N, датируемое 1994 и 1923 гг. Скорость эволюции данных геновариантов была наиболее высокой и составила 1,298 × 10–4 и 7,868 × 10–4 нуклеотидных замен/сайт/год. Выявлены рекомбинации в нижегородских последовательностях Med–, B95-8, China 1 с мутациями G212S, G212S, Е214Q соответственно.
Заключение. Впервые дана филодинамическая характеристика нижегородских изолятов и геновариантов LMP-1 ВЭБ, изолированных в разных регионах мира. Полученные данные расширяют существующие представления о циркуляции геновариантов LMP-1 ВЭБ на территории европейской части России.
369-379
ОБЗОРЫ
Молекулярно-генетический портрет вирулентности Stenotrophomonas maltophilia
Аннотация
Введение. Stenotrophomonas maltophilia является условно-патогенным микроорганизмом, обладающим природной устойчивостью к широкому спектру антибиотиков. Бактерия ассоциирована с рядом серьёзных заболеваний и вносит значимый вклад в патогенез полимикробных инфекций. S. maltophilia обладает широким набором факторов вирулентности, информация о которых к настоящему времени представлена в виде разрозненных и необобщённых данных.
Цели и задачи: критически проанализировать и обобщить актуальные данные, затрагивающие молекулярно-генетические аспекты вирулентности S. maltophilia, для более глубокого понимания патогенеза инфекций, связанных с этим возбудителем.
Материалы и методы. Выполнен анализ информации из 80 современных литературных источников, посвящённых изучению вирулентных свойств S. maltophilia на молекулярно-генетическом уровне. Анализ сфокусирован на механизмах продукции факторов вирулентности и определяющих их генетических детерминантах.
Результаты. Проанализированы и обобщены молекулярные механизмы вирулентности, детерминирующие вызванный S. maltophilia инфекционный процесс, включая адгезивную функцию поверхностных структур бактериальной клетки (липополисахариды, пили/фимбрии, флагеллы), продукцию внеклеточных энзимов, способность формировать биоплёнки на абиотических поверхностях и на тканях макроорганизма, фукционирование эффлюкс-помп, секрецию во внешнюю среду малых молекул системой межклеточного обмена информацией Quorum Sensing, а также влияние метаболизма железа на вирулентные свойства S. maltophilia.
Заключение. Адаптационные механизмы, позволяющие S. maltophilia приспосабливаться к новым нишам обитания, выживать в организме человека и неблагоприятных условиях окружающей среды, изучены недостаточно. Аналитический обзор, обобщающий актуальные сведения о молекулярно-генетических аспектах вирулентности S. maltophilia, будет интересен клиническим специалистам и исследователям, изучающим фундаментальные механизмы вирулентности.
380-390
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.