


Phylodynamic characteristics of the LMP-1 gene of the Epstein–Barr virus isolated in the Nizhny Novgorod region
- Authors: Bryzgalova D.A.1, Sakharnov N.A.1, Popkova M.I.1, Soboleva E.A.2, Kulova E.A.3, Utkin O.V.1
-
Affiliations:
- Academician I.N. Blokhina Nizhny Novgorod Research Institute of Epidemiology and Microbiology
- Nizhny Novgorod Regional Center for the Prevention and Control of AIDS and Infectious Diseases
- Tonus Krokha i Semeynaya Stomatologiya LLC
- Issue: Vol 100, No 5 (2023)
- Pages: 369-379
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18484
- DOI: https://doi.org/10.36233/0372-9311-379
- EDN: https://elibrary.ru/tqghak
- ID: 18484

Cite item
Full Text

Abstract
Introduction. Epstein–Barr virus (EBV) is one of the most common herpesviruses and has a pronounced genetic polymorphism. The study of the phylodynamic characteristics of the virus is an important aspect of the study of evolutionary changes in the LMP-1 gene and their consequences.
The aim of the work was a philodynamic analysis of EBV isolates from Nizhny Novgorod region based on the C-terminal fragment of the LMP-1 gene.
Materials and methods. The study included 158 EBV isolates obtained from blood leukocytes and saliva of children aged 1–17 years with a diagnosis of infectious mononucleosis caused by EBV (n = 68) and apparently healthy children of comparable sex and age (n = 29). LMP-1 genovariants were obtained using the Sanger sequencing method. Comparative analysis of amino acid sequences was performed using the MEGA X program. Philodynamic analysis of the obtained nucleotide sequences and isolates deposited in GenBank was carried out using the BEAST v. 1.10.4 software package. Recombination analysis was performed using the Simplot program.
Results. 158 nucleotide sequences of the C-terminal fragment of the LMP-1 gene from Nizhny Novgorod region EBV isolates were obtained and deposited in the GenBank database. The circulation time of the nearest common ancestor for the modified B95-8 genovariants with G212S + E328Q + S366T and NC mutations with the D250N substitution has been established dating back to 1994 and 1923. The rate of evolution of these genovariants was the highest and amounted to 1.298 × 10–4 and 7.868 × 10–4 nucleotide substitutions/site/year. Recombinations were detected in the Nizhny Novgorod region sequences Med-, B95-8, China 1 with mutations G212S, G212S, E214Q, respectively.
Conclusion. For the first time, a phylodynamic characterization of Nizhny Novgorod region isolates and LMP-1 EBV genovariants isolated in various regions of the world is given. The data obtained expand the existing understanding of the circulation of EBV LMP-1 genovariants in the territory of the European part of Russia.
Full Text
Введение
Вирус Эпштейна–Барр (ВЭБ) является представителем семейства Herpesviridae, подсемейства Gammaherpesvirinae, рода Lymphocryptoviruses, вида Human gammaherpesvirus 4. ВЭБ характеризуется генетическим разнообразием, которое обусловлено длительной эволюцией, включающей ряд аспектов. Показано, что рекомбинация играет определяющую роль в формировании разнообразия ВЭБ и структуры его генома [1]. Зафиксированы признаки диверсифицирующего отбора на уровне генов латентной фазы, функция которых заключается в переходе активной инфекции ВЭБ к длительной персистенции [2].
Одним из них является ген латентного мембранного белка 1 (LMP-1), характеризующийся наиболее высокой степенью генетической изменчивости по сравнению с другими вирусными генами. Большинство работ по изучению эволюции гена LMP-1 выполнено зарубежными исследователями. До настоящего времени в России исследования, посвящённые эволюционным изменениям LMP-1, основывались на филогенетическом анализе С-концевого фрагмента гена, изолированного в популяции древних народов (славян и татар), а также референсных последовательностей геновариантов в рамках классификации R. Edwards и соавт. [3, 4]. Данная классификация является наиболее распространённой и включает 7 геновариантов LMP-1: Alaskan, China 1, China 2, China 3, B95-8, Mediterranean (Med) и North Carolina (NC) [3]. Одним из важных аспектов классификации R. Edwards и соавт. является анализ вариаций наиболее полиморфного и охарактеризованного в структурно-функциональном плане С-концевого фрагмента гена LMP-1 [3]. Рядом исследователей были выявлены новые варианты гена LMP-1 (вне классификации R. Edwards и соавт.), такие как Southeast Asia 1 и 2 (SEA 1 и SEA 2), изолированные в Таиланде [5], Srb1 и Srb2 — в Сербии [6], а российскими учёными среди взрослого населения обнаружен уникальный древний вариант LMP1-TatK [4]. У инфицированных лиц во время репликации ВЭБ в его геноме возникают мутации, которые приводят к генетическому разнообразию вируса [7]. Данные спонтанные замены, накопленные у взрослых инфицированных лиц, не всегда закрепляются в популяции, но могут влиять на конечный результат при оценке эволюционных изменений геновариантов LMP-1. Поэтому для исследования распространённости и эволюции С-концевого фрагмента гена LMP-1 мы использовали последовательности, полученные у детей. Несмотря на значительное количество работ по изучению генетического разнообразия LMP-1, эволюционные изменения гена и их последствия изучены недостаточно.
В современный период широкое применение получил филодинамический анализ — метод оценки эволюции патогенов с учётом временны́х параметров. Основными филодинамическими параметрами являются скорость эволюции и время циркуляции ближайшего общего предка. Данный метод позволяет оценить динамику и направление эволюционных изменений различных патогенов.
Цель исследования — филодинамический анализ нижегородских изолятов ВЭБ на основе C-концевого фрагмента гена LMP-1.
Материалы и методы
Исследовали 158 изолятов ВЭБ, полученных из лейкоцитов крови и слюны 68 детей 1–17 лет, госпитализированных в Детскую инфекционную больницу № 8 г. Нижнего Новгорода с диагнозом «инфекционный мононуклеоз, вызванный ВЭБ», и 29 условно здоровых детей сопоставимого пола и возраста, проходивших диспансеризацию в ООО «Тонус Кроха и семейная стоматология» (Нижний Новгород). У законных представителей несовершеннолетних пациентов получено добровольное информированное согласие в соответствии с положениями Хельсинкской декларации (2013). Протокол исследования одобрен локальным этическим комитетом Нижегородского НИИ эпидемиологиии микробиологии им. академика И.Н. Блохиной (протокол № 3 от 11.11.2021).
Фракцию лейкоцитов получали с помощью реагента «Гемолитик» (ЦНИИ Эпидемиологии Роспотребнадзора). Пробоподготовку слюны проводили по оптимизированному нами способу, изложенному ранее [9]. Экстракцию тотальной нуклеиновой кислоты из лейкоцитов крови и слюны производили при помощи комплекта реагентов «РИБО-преп» (ЦНИИ Эпидемиологии Роспотребнадзора) с модификациями [9, 10]. Наличие вирусной ДНК ВЭБ подтверждали методом полимеразной цепной реакции (ПЦР) в реальном времени с применением набора реагентов «АмплиСенс EBV/CMV/HHV6- cкрин-FL» (ЦНИИ Эпидемиологии Роспотребнадзора) на амплификаторе «Rotor-Gene Q 5plex HRM» («Qiagen»).
С-концевой фрагмент гена LMP-1 амплифицировали с помощью разработанного нами лабораторного протокола, базирующегося на использовании метода ПЦР с праймерами, описанными в статье [11]. Одностадийный вариант ПЦР проводили, используя праймеры: А1 5’-AGT CAT AGT AGC TTA GCT GAA-3’и A2 5’-CCA TGG ACA ACG ACA CAG T-3. Размер фрагмента С-концевой области гена LMP-1 — 602 п.н.
Секвенирование очищенных амплифицированных фрагментов ДНК проводили с помощью набора реагентов «Big Dye Terminator v.3.1 Cycle Sequencing Kit» («Applied Biosystems») на генетическом анализаторе «AB-3500 Genetic Analyzer» («Applied Biosystems») с использованием оригинального программного обеспечения «3500 Data Collection Software v. 1.0» («Applied Biosystems»).
Нуклеотидные последовательности анализировали с помощью открытого программного обеспечения «MEGA X» («Mega Software»)1.
Формирование выборки
Для изучения разнообразия C-концевого фрагмента гена LMP-1 ВЭБ были проанализированы доступные в базе данных GenBank нуклеотидные последовательности размером более 80% открытой рамки считывания. В выборку вошли 763 нуклеотидные последовательности гена LMP-1 и генома ВЭБ, изолированные после 1970 г. на территории Австралии, Аляски, Аргентины, Бразилии, Великобритании, Ганы, Гонконга, Индонезии, Кении, Китая, Кореи, Нигерии, Пакистана, Польши, Сербии, США, Тайваня, Таиланда, Хорватии, Швейцарии, Японии. Последовательности гена LMP-1, полученные другими отечественными исследователями на территории России, в указанной базе данных не представлены.
Филодинамический анализ
Выравнивание нуклеотидных последовательностей наряду с формированием аминокислотного кода LMP-1 проводили с помощью программного обеспечения «MEGA X» [12]. Для построения филогенетического дерева использовали метод максимального правдоподобия, пакет программ «BEAST v. 1.10.4» [8]. Для анализа полученных нуклеотидных последовательностей была подобрана модель Hasegawa–Kishino–Yano. На основе модели SkyGrid рассчитывали динамику демографических показателей [13]. Достоверными считали уровни апостериорной вероятности > 0,85. На базе строгих молекулярных часов измеряли скорость эволюции. Длина цепи Маркова Монте-Карло составила 200 млн шагов. Графическую визуализацию дерева осуществляли в программе «FigTree 1.4.3». Дендрограммы анализировали с помощью программы «Tracer v. 1.7.1» [14].
По данным российских авторов, одним из типов генетической изменчивости, которые способствуют эволюции гена LMP-1, являются точечные мутации [4]. В связи с этим нами проведён сравнительный анализ точечных аминокислотных замен в последовательностях исследуемой выборки с целью определения специфических мутаций, характерных для определённых регионов, а также отслеживания их распространения со временем.
Рекомбинационный анализ
Предполагаемые рекомбинантные последовательности анализировали с помощью программного обеспечения «Simplot» [15] по методу попарного внутригруппового невзвешенного среднего (unweighted pair group method with arithmetic mean, UPGMA) с длиной окна 200 п.н., шаг 20 п.н. и 100 повторениями bootstrap-анализа. Для анализа нуклеотидных последовательностей использовали модель TN93 (Tamura-Nei, 93).
Результаты
На первом этапе работы нами получены и депонированы в базу данных GenBank 158 нуклеотидных последовательностей С-концевого фрагмента гена LMP-1 нижегородских изолятов ВЭБ (регистрационные номера OP105219-OP105376 присвоены 09.08.2022).
Филогенетический анализ
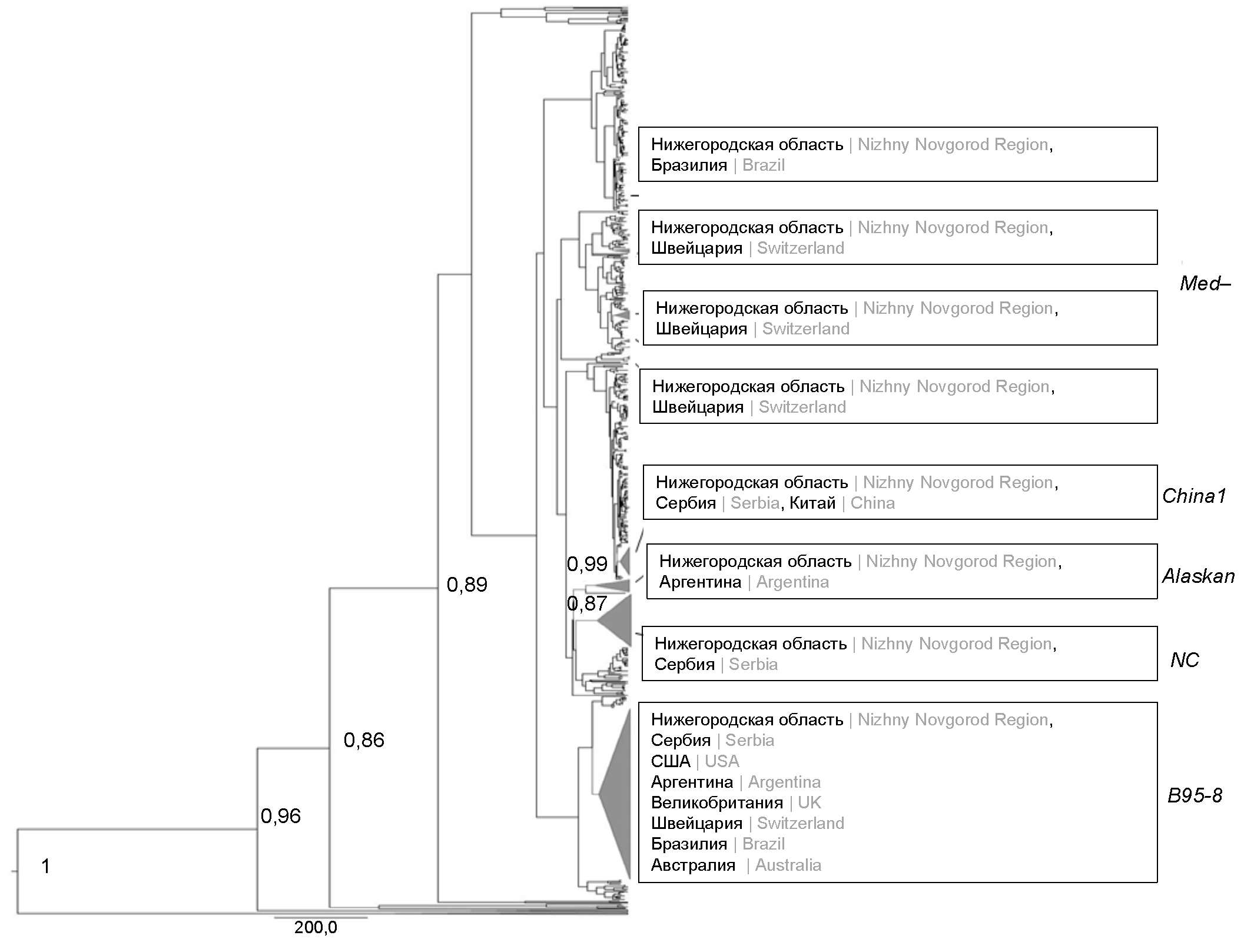
Для оценки кластеризации нижегородских изолятов ВЭБ, а также характера их взаимоотношений с последовательностями, изолированными в 1970–2022 гг. в различных регионах мира, было сконструировано филогенетическое дерево (рис. 1).

Рис. 1. Байесово филогенетическое дерево MCC, сконструированное на основе нуклеотидных последовательностей C-концевого фрагмента гена LMP-1 нижегородских изолятов ВЭБ и последовательностей, полученных в различных регионах мира и доступных в GenBank. MCC — филогенетическое дерево с максимальной надёжностью клад. Показаны байесовские апостериорные вероятности более 0,85 в основных узлах. Треугольниками обозначены клады с нижегородскими изолятами ВЭБ и соседними последовательностями, доступными в GenBank.
Только один идентифицированный нами нижегородский изолят (OP105219) образовывал единый кластер и показал высокую вероятность филогеографической связи (значение апостериорной вероятности, равной 0,99) с референсной последовательностью Alaskan и последовательностями, полученными в Аргентине. Общую группу также образовывали изоляты China 1, полученные из Нижегородской области, Сербии и Китая. Однако значительного генетического сходства между ними не выявлено в связи с низкими значениями апостериорной вероятности. Похожая апостериорная вероятность отмечалась и в кластере с нижегородскими изолятами B95-8. Эта группа включала наибольшее количество последовательностей, выделенных в различных регионах мира (Сербии, Аргентине, Великобритании, Швейцарии, Бразилии, Австралии и США). Генетическое родство с апостериорной вероятностью 0,87 с последовательностями из Сербии показали нижегородские изоляты ВЭБ и геновариант NC, сформировав на дереве единую монофилетическую группу. Нижегородские последовательности Med– не образовывали отдельного единого кластера, а распределились между генетически близкими изолятами из Бразилии и Швейцарии.
Скорость эволюции
Скорость эволюции C-концевого фрагмента гена LMP-1 ВЭБ в кластерах, содержащих нижегородские изоляты China 1 и Alaskan, варьировала незначительно и составила 5,042 × 10–5 и 1,518 × 10–5 нуклеотидных замен/сайт/год. В кластерах с последовательностями из Нижегородской области NC и B95-8 скорость эволюции была несколько выше — 1,298 × 10–4 и 7,868 × 10–4 нуклеотидных замен/сайт/год. Для всей исследуемой выборки C-концевого фрагмента гена LMP-1 ВЭБ скорость эволюции составила 2,32 × 10–4 нуклеотидных замен/сайт/год.
Время циркуляции ближайшего общего предка
Клада, содержащая нижегородские изоляты B95-8 и последовательности из Сербии, Аргентины, Великобритании, Швейцарии, Бразилии, Австралии и США, включала общую комбинацию мутаций G212S + E328Q + S366T. Нами было определено время циркуляции ближайшего общего предка для данного геноварианта B95-8 с заменами G212S + E328Q + S366T, которое датируется 1994 г. Клада, содержащая нижегородские изоляты NC и последовательности из Сербии, включает общую замену D250N и предположительно имеет общего предка, время циркуляции ближайшего общего предка — 1923 г.
Сравнительный анализ аминокислот
На следующем этапе работы нами проведён сравнительный анализ данных точечных аминокислотных замен в последовательностях, доступных в GenBank и выделенных в различных географических регионах мира. Также производился поиск других общих аминокислотных мутаций в последовательностях геновариантов Med, China 1, Alaskan. Анализ включал изоляты ВЭБ, ранее охарактеризованные по классификации R. Edwards и соавт. в других исследованиях [6, 16–22]. Масштабные исследования, посвящённые данному вопросу, проводились только на территориях Аргентины, Бразилии, Сербии, Китая, Швейцарии, Америки.
Сравнительный анализ точечных аминокислотных замен в последовательностях исследуемой выборки продемонстрировал высокую распространённость мутаций, не характерных для определённых геновариантов (таблица).
Сравнение частоты распространения геновариантов LMP-1 ВЭБ в мире
Comparison of the prevalence of EBV LMP-1 genovariants in the world
Регион Region | B95-8 | Med (G212S) | NC (D250N) | China 1 (E214Q) | |
G212S + E328Q + S366T | G212S + E328Q | ||||
Нижегородская область Nizhny Novgorod Region | 99 % (95/96) | – | 78% (7/9) | 96% (27/28) | 50% (12/24) |
Сербия | Serbia | – | 100% (23/23) | 77% (14/18) | 100% (10/10) | – |
США | USA | 80% (8/10) | – | – | – | – |
Бразилия | Brazil | 50 % (3/6) | – | 70% (19/27) | – | – |
Аргентина | Argentina | 87% (7/8) | – | 94% (34/36) | – | 79% (23/29) |
Иран | Iran | – | 100% (4/4) | 59% (13/22) | – | – |
Швейцария | Switzerland | 75% (3/4) | – | – | – | – |
Самая малочисленная группа, состоящая из 11 последовательностей, принадлежала геноварианту LMP-1 Alaskan, который редко встречается во всём мире. Проанализированные изоляты не выявили общих аминокислотных замен, помимо мутаций, охарактеризованных R. Edwards и соавт. [3].
Для геноварианта NC была характерна мутация D250N, которая встречалась только в изолятах из Нижегородской области и Сербии в 94,4–100% случаев соответственно. Как видно из таблицы, наиболее широко распространёнными в различных регионах мира были мутации G212S + E328Q + S366T в изолятах B95-8 (50–99%) и G212S в Med (59–94%).
Ещё одной мутацией, обнаруженной в нашем исследовании, является E214Q, которая была характерна для геноварианта China 1 и встречалась в нижегородских изолятах и в последовательностях из Аргентины с частотой 46 и 79% соответственно (таблица). Мы предполагаем, что мутация E214Q появилась в нижегородских изолятах China 1 в результате их рекомбинации с последовательностью штамма Raji. По аналогии мутация G212S появилась в изолятах Med и B95-8 в результате их рекомбинации с последовательностью CAO, т.к. данная замена характерна для данного штамма. Похожая ситуация прослеживается и для мутации S366T в B95-8. В связи с этим нами был проведён рекомбинационный анализ данных мутаций при помощи бутскан-анализа.
Рекомбинационный анализ
Для определения возможных событий рекомбинации все нижегородские последовательности B95-8 с заменами G212S + S366T тестировали в сравнении с референсными последовательностями CАО и B95-8. Референсная последовательность Alaskan использовалась в качестве внешней группы. Точка рекомбинации (R1) была обнаружена во всех последовательностях в домене CTAR1 (192–232 а.к.), ответственного за активацию транскрипционного фактора NF-κB (рис. 2, а) [23]. Этот результат указывает на то, что мутация G212S в изолятах B95-8 действительно могла появиться в процессе рекомбинации между последовательностями B95-8 и CАО.

Рис. 2. Рекомбинационный анализ нижегородских последовательностей. а — бутскан-анализ нижегородских последовательностей B95-8 с мутациями G212S + S366T относительно референсных последовательностей CАО, B95-8 и Alaskan (внешняя группа); б — бутскан-анализ нижегородских последовательностей с мутацией Е214Q относительно последовательностей Raji, China 1 и CАО (внешняя группа); в — бутскан-анализ нижегородских последовательностей Med– с мутацией G212S относительно последовательностей Med–, CAO и B95-8 (внешняя группа). Параметры анализа: окно 200 п.н., шаг 20 п.н. и 100 повторений по методу UPGMA.
Мы также проанализировали все нижегородские последовательности, расположенные в одном кластере вместе с China 1 и содержащие мутацию Е214Q, которая характерна для штамма Raji. При тестировании против референсных последовательностей Raji, China 1 и CАО, вариант China 1 с мутацией Е214Q показал точку рекомбинации (R2) в области домена CTAR1 (192–232 а.к.) во всех последовательностях (рис. 2, б). Таким образом, можно говорить о том, что мутация Е214Q в нижегородских изолятах China 1 появилась в результате рекомбинации последовательностей China 1 и Raji.
В дальнейшем нами проведён рекомбинационный анализ всех нижегородских последовательностей, которые образовали кладу вместе с Med– и содержали мутацию G212S, характерную для штамма CAO. В качестве внешней группы была включена последовательность B95-8. На рис. 2, в видна общая область (R3) пересечения всех нижегородских последовательностей Med– с мутацией G212S и CAO в домене CTAR1 (192–232 а.к.). В связи с этим мутация G212S в нижегородских последовательностях Med– могла появиться вследствие рекомбинации Med– и CAO.
Обсуждение
В настоящей работе впервые дана филодинамическая характеристика нижегородских изолятов ВЭБ, полученных от детей с диагнозом «инфекционный мононуклеоз, вызванный ВЭБ» и здоровых доноров, и геновариантов LMP-1 ВЭБ, изолированных в различных регионах мира. Для оценки эволюционных взаимоотношений между последовательностями С-концевого фрагмента гена LMP-1 ВЭБ в исследуемой выборке мы реконструировали филогенетическое дерево с применением байесовской статистики. Филогенетический анализ на основе нуклеотидных последовательностей С-концевого фрагмента гена LMP-1 показал, что нижегородские изоляты, относящиеся к геноварианту NC, генетически близки с последовательностями из Сербии. Это можно объяснить активными миграционными процессами, происходящими вследствие исторически сложившихся межгосударственных взаимоотношений данных стран. Сравнительный анализ аминокислотных замен нижегородских изолятов и последовательностей из Сербии, отнесённых к геноварианту NC, продемонстрировал наличие общей мутации D250N для данных регионов. Время происхождения ближайшего общего предка для геноварианта NC с наличием мутации D250N изолятов из Сербии и Нижегородской области датируется 1923 г., что соотносится с периодом русской эмиграции во время гражданской войны в Королевство Югославию, в которую входили Сербия, Хорватия и Словения. Больше всего русских переселенцев было размещено в Сербии, где насчитывалось около 200 колоний [24]. В настоящей момент функциональная значимость замены D250N для геноварианта NC не определена.
Особый интерес для нас представляла клада, содержащая нижегородские последовательности B95-8 и изоляты, полученные из Сербии, Аргентины, Великобритании, Швейцарии, Бразилии, Австралии и США. Несмотря на то что апостериорная вероятность для данной клады была невысокой, почти все последовательности включали комбинацию из 3 аминокислотных замен G212S + S366T + E328Q и были изолированы после 1999 г. Исключение составляли изоляты из Сербии, в которых сравнение комбинации мутаций G212S + S366T + E328Q было невозможным из-за методических ограничений, т.к. данные последовательности, размещённые в GenBank, не содержали исследуемый отрезок с заменой S366T. Стоит обратить внимание на то, что время происхождения общего ближайшего предка для клады B95-8 с мутациями G212S + S366T + E328Q датируется 1994 г. Проведённый нами сравнительный анализ точечных аминокислотных мутаций G212S + S366T + E328Q геноварианта B95-8 показал их высокую распространённость в последовательностях, полученных в различных регионах мира после 1999 г. Отметим, что высокая распространённость модифицированного варианта B95-8 с заменами G212S + S366T + E328Q не связана с определёнными территориями, а встречается с высокой частотой в последовательностях, изолированных в различных регионах мира. Модифицированный вариант B95-8 с мутациями G212S + S366T + E328Q преобладал в Бразилии, Аргентине, Иране. Похожая картина наблюдалась в последовательностях из Ирана, в которых мутации G212S + E328Q в изолятах B95-8 были широко распространены, а замена S366T не учитывалась. Данные последовательности не были депонированы в GenBank, и расчёт был произведён на основе материалов статьи B. Sarshari и соавт. [22]. Распространённость геноварианта B95-8 на данных территориях была невысокой по сравнению с другими геновариантами LMP-1. Другая картина представлена в Евразии, где B95-8 является часто встречающимся геновариантом в большинстве стран. При анализе имеющихся последовательностей в GenBank выявлено, что совокупность мутаций G212S + E328Q была характерна для всех изолятов из Сербии, относящихся к B95-8. Примечательно, что мутация E328Q, по данным российских авторов, не типична для европейских образцов, а широко распространена среди населения Дальнего Востока России [25]. В нижегородских изолятах мутации G212S + S366T + E328Q встречались в абсолютном большинстве последовательностей B95-8. Мы полагаем, что после 1999 г. в мире произошла смена циркуляции прототипного геноварианта B95-8 на модифицированный вариант с мутациями G212S + E328Q + S366T. Наши предположения подтверждаются и отсутствием информации о данных мутациях в геноварианте B95-8 в одном из масштабных исследований 1999 г. [3]. Нами также было показано, что замена G212S в модифицированном геноварианте B95-8 является результатом рекомбинации с последовательностью CAO.
Широкое распространение последовательностей B95-8 с мутациями G212S + S366T + E328Q может быть связано с тенденцией к росту числа заболеваний, ассоциированных с ВЭБ. По данным российских авторов, рост заболеваемости инфекционным мононуклеозом наблюдается как в целом по России, так и в Нижегородской области [26]. Растёт число случаев, требующих госпитализации [27].
Данная тенденция может быть связана с влиянием замен G212S + S366T + E328Q на функциональную активность белка LMP-1. Замены G212S и S366T ассоциированы с геновариантом LMP-1 CAO. Известно, что геновариант CAO обладает большей трансформационной активностью, чем геновариант B95-8 [25]. Существуют исследования, сосредоточенные на изучении влияния мутаций G212S + S366T на функциональную активность геноварианта B95-8 в клеточных линиях. Так, в работе отечественных авторов было показано, что геновариант B95-8 с заменами G212S + S366T усиливает активацию транскрипционного фактора NF-κB, который играет важную роль в регуляции ключевых функций клетки, таких как рост и выживаемость, старение и опухолевая трансформация [23]. В другой работе показано, что штамм B95-8 при наличии мутации G212S + S366T демонстрировал сниженную или утраченную способность активировать микроРНК (miR-155 и miR-193b), которые играют важную роль в различных физиологических и патологических процессах [28]. Влияние комбинации мутаций G212S + E328Q + S366T на функциональную активность белка LMP-1 B95-8 ещё предстоит изучить.
Интересно, что оба геноварианта B95-8 с мутациями G212S + E328Q + S366T и NC с заменой D250N имели скорость эволюции выше, чем у остальных геновариантов. Данный факт может объяснять высокую распространённость геновариантов B95-8 с мутациями G212S + E328Q + S366T и NC с заменой D250N в определённых регионах мира. Нами были получены данные, характеризующие скорость эволюции для всей исследуемой выборки С-концевого фрагмента гена LMP-1 ВЭБ. Скорость эволюции 2,32 × 10–4 нуклеотидных замен/сайт/год была несколько выше по сравнению со значениями, полученными для гена LMP-1 другими исследователями из Аргентины и Бразилии (8,591 × 10–5 и 3,8 × 10–5 замен/сайт/год соответственно). Это может быть связано с тем, что итоговые результаты скорости эволюции определяются размером выборки и её составом.
Отметим, что по результатам нашего исследования геноварианты B95-8, а также Med–/Med+, несущие аминокислотную замену G212S, и China 1 с мутацией E214Q отличались наличием рекомбинации с другими штаммами и геновариантами вируса и характеризовались тенденцией к широкому распространению в популяции, что может в дальнейшем оказывать влияние на эволюцию гена LMP-1. В связи с этим остановимся на характеристике указанных мутаций.
Геноварианты Med– и Med+ с заменой G212S чаще обнаруживали в последовательностях, полученных из разных регионов мира, изолированных после 1999 г. С высокой частотой данная мутация встречалась в изолятах Med, выделенных на территории Аргентины, Бразилии, Сербии, Ирана, а также в исследованных нами изолятах. Полученные нами результаты рекомбинационного анализа указывают на то, что мутация G212S также могла возникнуть в геноварианте Med– в результате рекомбинации с последовательностью штамма CAO. На филогенетическом дереве нижегородские изоляты Med– не образовывали единой клады. Это связано с тем, что данный геновариант относится к высокодивергентным.
Ранее в российских исследованиях распространённость мутаций G212S, S366T, E328Q не соотносили с определёнными геновариантами классификации R. Edwards и соавт. [25, 29].
Менее распространённой была мутация E214Q, характерная для геноварианта China 1. Ранее замена E214Q была охарактеризована в отношении аргентинского геноварианта China 1* и встречалась независимо от групп исследования в 79% (23/29) изолятов [20]. Мутацию E214Q M. Gantuz и соавт. описывали как результат рекомбинации между аргентинским геновариантом China 1* и штаммом Raji, содержащим эту замену в референсной последовательности, и утверждали, что высокая распространённость China 1* может быть обусловлена генетическими и/или иммунологическими факторами [20]. Мутация E214Q встречалась также в нижегородских изолятах China 1 в 50% (12/24) изолятов. В исследованиях других российских авторов, проведённых ранее, данные о распространённости мутации E214Q в изолятах China 1 отсутствуют. Проведённый нами рекомбинационный анализ показал, что мутация E214Q в нижегородских изолятах China 1 появилась в результате рекомбинации со штаммом Raji. Мутация E214Q расположена в DSGxxS — каноническом мотиве (HOS-сайт). Этот сайт входит в состав E3-лигазы, которая участвует в процессинге молекулы IkB, являющейся супрессором NF-κB [25]. Функциональная значимость замены E214Q для геноварианта China 1 требует дальнейшего изучения.
Таким образом, анализ аминокислотных последовательностей LMP-1 геновариантов B95-8, NC, Med и China 1, полученных в различных регионах мира, выявил ряд специфических мутаций, которые при определённых условиях (снижении иммунитета, воздействии неблагоприятных факторов окружающей среды) могут оказывать влияние на развитие и характер течения ВЭБ-ассоциированных заболеваний, в том числе инфекционного мононуклеоза.
Наше исследование, а также работа М. Gantuz и соавт. [20] позволяют взглянуть с другой стороны на утверждение о том, что C-концевая область гена изменялась без какой-либо истории рекомбинации по сравнению с N-концом и трансмембранной областью LMP-1, о чём упоминалось ранее в работе J.M. Burrows и соавт. [30]. Появление большого разнообразия новых мутаций, специфичных для определённых геновариантов, может быть следствием рекомбинации, что подчёркивает важность этого механизма для возникновения новых вариантов вируса.
Заключение
Впервые в России нами проведён филодинамический анализ последовательностей С-концевой области гена LMP-1 ВЭБ, изолированных у детей с инфекционным мононуклеозом и здоровых вирусоносителей.
Показано, что скорость эволюции нижегородских изолятов B95-8 с набором мутаций G212S + E328Q + S366T и изолятов NC с мутацией D250N была выше по сравнению с описанными ранее в других регионах мира, также для них было определено время циркуляции общего предка (1994 и 1923 гг. соответственно).
Среди нижегородских изолятов выявлено преобладание последовательностей, содержащих штамм-неспецифичные аминокислотные замены G212S (в геновариантах B95-8 и Med–) и Е214Q (в геноварианте China 1). При этом они характеризовались наличием рекомбинаций с другими штаммами, что может в перспективе способствовать формированию новых ветвей эволюции гена LMP-1.
Выявленные нами молекулярно-генетические характеристики изолятов LMP-1 отражают особенности эволюции ВЭБ в Нижегородской области (относится к европейской части России) и формирования его генетического разнообразия. При этом отмечается направленность эволюции гена LMP-1 в сторону увеличения частоты мутаций, ассоциированных с повышенной трансформирующей активностью вируса.
Полученные результаты являются основой для перспективных исследований фенотипических свойств выявленных мутаций LMP-1, оценки их клинической значимости и взаимосвязи с особенностями эпидемического процесса ВЭБ-инфекции.
1 URL: https://www.megasoftware.net
About the authors
Daria A. Bryzgalova
Academician I.N. Blokhina Nizhny Novgorod Research Institute of Epidemiology and Microbiology
Email: saharnov@nniiem.ru
ORCID iD: 0000-0002-6663-8440
junior researcher, Laboratory of molecular biology and biotechnology, Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Russian Federation, Nizhny NovgorodNikolay A. Sakharnov
Academician I.N. Blokhina Nizhny Novgorod Research Institute of Epidemiology and Microbiology
Author for correspondence.
Email: saharnov@nniiem.ru
ORCID iD: 0000-0003-3965-2033
Cand. Sci. (Biol.), senior researcher, Laboratory of molecular biology and biotechnology
Russian Federation, Nizhny NovgorodMaria I. Popkova
Academician I.N. Blokhina Nizhny Novgorod Research Institute of Epidemiology and Microbiology
Email: saharnov@nniiem.ru
ORCID iD: 0000-0001-5864-5862
Cand. Sci. (Мed.), leading researcher, Laboratory of molecular biology and biotechnology, Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Russian Federation, Nizhny NovgorodEvgeniya A. Soboleva
Nizhny Novgorod Regional Center for the Prevention and Control of AIDS and Infectious Diseases
Email: saharnov@nniiem.ru
ORCID iD: 0000-0003-3591-9618
infectious disease physician, Nizhny Novgorod Regional Center for the Prevention and Control of AIDS and Infectious Diseases
Russian Federation, Nizhny NovgorodEkaterina A. Kulova
Tonus Krokha i Semeynaya Stomatologiya LLC
Email: saharnov@nniiem.ru
ORCID iD: 0000-0002-5207-1164
Cand. Sci. (Мed.), infectious disease physician, allergologist and immunologist, Tonus Krokha i Semeynaya Stomatologiya LLC
Russian Federation, Nizhny NovgorodOleg V. Utkin
Academician I.N. Blokhina Nizhny Novgorod Research Institute of Epidemiology and Microbiology
Email: saharnov@nniiem.ru
ORCID iD: 0000-0002-7571-525X
Cand. Sci. (Biol.), Head, Laboratory of molecular biology and biotechnology, Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Russian Federation, Nizhny NovgorodReferences
Supplementary files

