


Том 102, № 3 (2025)

- Год: 2025
- Выпуск опубликован: 15.07.2025
- Статей: 13
- URL: https://microbiol.crie.ru/jour/issue/view/190

ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
Актуализация данных о сибиреязвенных стационарно неблагополучных пунктах и почвенных очагах как основа совершенствования эпизоотолого-эпидемиологического мониторинга сибирской язвы в Российской Федерации

Аннотация
Введение. Сибирская язва в Российской Федерации регистрируется ежегодно. Сохранение постоянного риска осложнения эпизоотолого-эпидемиологической ситуации по сибирской язве обусловлено повсеместным распространением почвенных очагов инфекции (сибиреязвенных захоронений (СЯЗ), «моровых полей») и связанных с ними стационарно неблагополучных пунктов (СНП).
Цель работы — актуализация данных о сибиреязвенных СНП и почвенных очагах с целью совершенствования эпидемиологического надзора за сибирской язвой в России.
Материалы и методы. Использованы архивные, справочные материалы о сибиреязвенных СНП и почвенных очагах, учётные и отчётные данные территориальных органов Роспотребнадзора и ветеринарной службы. Осуществлён подбор критериев характеристики сибиреязвенных СНП, СЯЗ и «моровых полей», с использованием которых разработана структура баз данных СНП и почвенных очагов сибирской язвы.
Результаты. Впервые разработаны электронные базы данных сибиреязвенных СНП и почвенных очагов на территории России, содержащие актуализированную информацию о характеристиках 32 566 СНП и 3314 почвенных очагов (3185 СЯЗ и 129 «моровых полей»). Анализ данных выявил снижение числа СНП и СЯЗ на территории страны по сравнению со справочными сведениями, а также отсутствие корреляции между учтённым количеством СНП и СЯЗ в большинстве регионов, что указывает на наличие большого числа неучтённых СЯЗ и сохранение потенциальных рисков осложнения ситуации по инфекции.
Заключение. Внедрение в практику органов и учреждений Роспотребнадзора, ветеринарной службы актуальных баз данных сибиреязвенных СНП и почвенных очагов позволит повысить уровень информационного обеспечения и эффективности эпидемиологического надзора за сибирской язвой в России.
 271-283
271-283



Филогения штаммов Yersinia pestis линии 4.ANT из Тувинского горного и сопредельных очагов чумы
Аннотация
Введение. Тувинский горный очаг чумы (ТГОЧ) в России с момента его открытия в 1964 г. проявляет постоянную эпизоотическую активность. Штаммы Yersinia pestis, выделяемые в этом очаге, относятся к филогенетической линии 4.ANT античного биовара основного подвида. Они высоковирулентны и эпидемически значимы. Использование современных молекулярно-генетических технологий позволит определить популяционную структуру штаммов 4.ANT в ТГОЧ.
Цель исследования — филогенетический и популяционный анализ штаммов Y. pestis линии 4.ANT из ТГОЧ по данным полногеномного SNP-типирования (single nucleotide polymorphism) и MLVA25-типирования (multiple locus variable number tandem repeats analysis).
Материалы и методы. Использованы полногеномные нуклеотидные последовательности 68 штаммов Y. pestis, включая 60 штаммов линии 4.ANT. Секвенирование штаммов проводили на платформе MGI. SNP-анализ выполняли путём выравнивания последовательностей в программе «Snippy v. 4.6» с последующим построением дендрограммы Maximum Likelihood на основе выявленных коровых SNPs в программе «SeaView». SNPs, маркерные для штаммов линии 4.ANT, выявляли при помощи программы «MEGA11». MLVA-генотипирование штаммов Y. pestis линии 4.ANT проводили путём поиска локусов с последующим подсчётом количества тандемных повторов в программе «Tandem Repeats Finder». Построение MLVA-дендрограммы выполняли методом UPGMA в программе «BioNumerics v. 7.6.3».
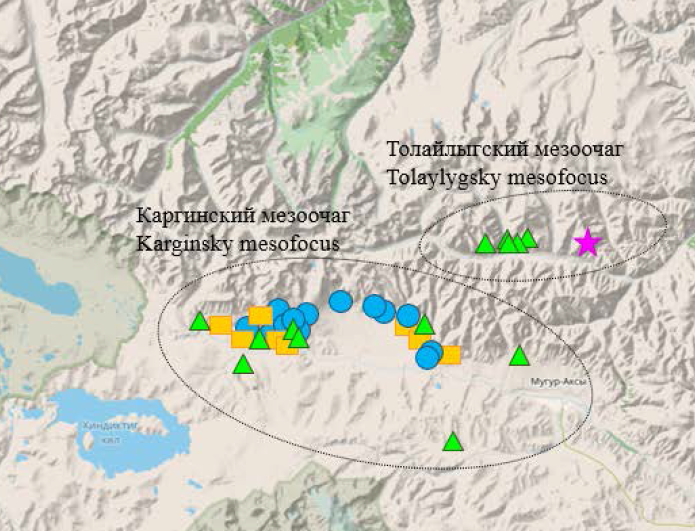
Результаты. По данным SNP-анализа штаммов Y. pestis линии 4.ANT из ТГОЧ установлено наличие 4 филогеографических групп: T1 (Саглинский, Толайлыгский и Барлыкский мезоочаги, 1971–1987 гг.), Т2 (Каргинский мезоочаг, 2014–2024 гг.), Т3 (Каргинский мезоочаг, 1977–2009 гг.), Т4 (Каргинский, Толайлыгский и Боро-Шайский мезоочаги, 2006–2013 гг.). Выявлены 8 MLVA-генотипов штаммов линии 4.ANT из Тувы и вариабельные VNTR-локусы: yp1290ms04, yp1935ms05, yp0559ms15, yp4042ms35, yp4425ms38, yp1108ms45, yp4280ms62, yp1580ms70.
Обсуждение. Среди штаммов, взятых в анализ, наиболее ранними представителями ветви 4.ANT выступают штаммы кластера Т1 из ТГОЧ. Отдельными подветвями на дереве представлены популяция штаммов из Горного Алтая и Монголии и популяция штаммов из ТГОЧ (1977–2024 гг.). Последняя популяция представлена политомией и характеризуется выраженной кластеризацией по пространственно-временнóму принципу.
Заключение. Определено наличие 4 основных филогеографических групп в популяции 4.ANT в ТГОЧ и установлены генетические различия между ними, что может быть использовано для углублённой молекулярно-генетической дифференциации и типирования штаммов Y. pestis в этом очаге.
284-295
Стабильность вакцинных штаммов сезонной живой гриппозной вакцины при их адаптации к культуре клеток MDCK
Аннотация
Введение. Подавляющее большинство гриппозных вакцин в мире производится с использованием развивающихся куриных эмбрионов (РКЭ) в качестве субстрата, однако активно обсуждается вопрос о переводе производства вакцин на перевиваемые клеточные линии, что обеспечит бесперебойность в условиях пандемии птичьего гриппа, а также позволит применять вакцину у лиц с аллергией на куриный белок. При накоплении вакцинных штаммов живой гриппозной вакцины (ЖГВ) в клетках млекопитающих могут возникать адаптационные мутации, влияющие на антигенные и иммуногенные свойства вакцины.
Цель работы — изучить биологические свойства вакцинных штаммов ЖГВ подтипов А/H1N1 и A/H3N2, полученных классическим способом в РКЭ, при их адаптации к культуре клеток почки собаки Мадина–Дарби (MDCK).
Материалы и методы. В работе были использованы штаммы для аттенуированной ЖГВ А/17/Калифорния/2009/38 (H1N1pdm09) и А/17/Техас/12/30 (H3N2). Мы провели серийное пассирование этих вирусов на MDCK и проанализировали ростовые свойства изолированных методом бляшек клонов in vitro и in vivo, их иммуногенность, перекрёстную реактивность и защитную эффективность на модели мышей, а также с использованием гипериммунных крысиных сывороток. Экспериментальные серии вакцинных штаммов ЖГВ А/17/Боливия/2013/6585 (H1N1), А/17/Швейцария/2013/1 (H3N2) и В/60/Пхукет/2013/26 были наработаны на культуре MDCK в ГНЦ ВБ «Вектор». Мы провели секвенирование генов поверхностных белков клеточных моновакцин и сравнили мутации, обнаруженные в гемагглютинине и нейраминидазе при адаптации к культуре клеток MDCK в лабораторных и производственных условиях.
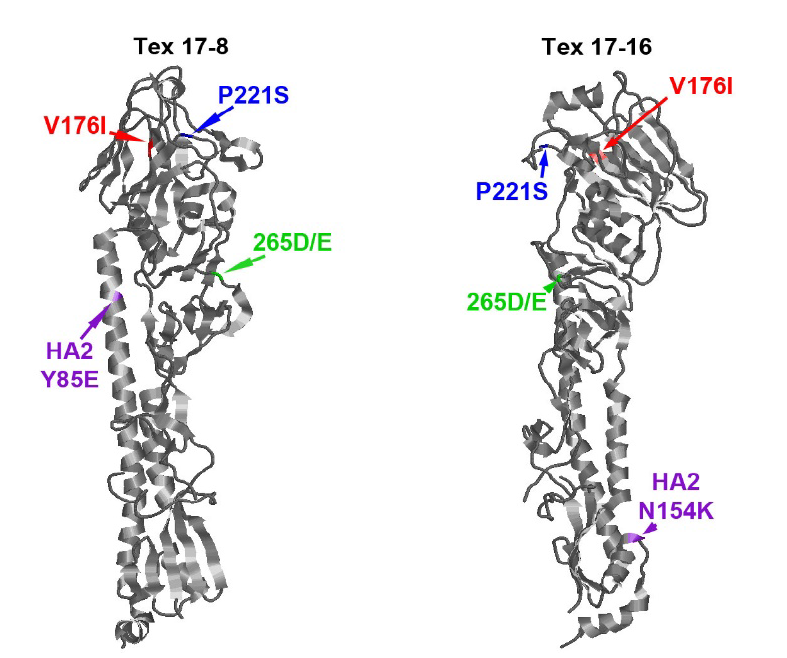
Результаты. Секвенирование поверхностных антигенов MDCK-адаптированных вариантов вируса A/H1N1 обнаружило адаптационные мутации в молекуле гемагглютинина — N156D (субъединица НА1) и A44V (субъединица НА2), одновременное присутствие которых усиливало репликативные свойства вакцинного штамма ЖГВ H1N1 в культуре клеток MDCK. Изучение данного адаптированного к культуре клеток MDCK варианта в эксперименте на мышах не выявило влияния обнаруженных мутаций на иммуногенные и протективные свойства вакцины. Адаптация вакцинного штамма ЖГВ H3N2 к культуре клеток MDCK привела к появлению существенно большего количества замен в молекуле НА, по сравнению с вирусом H1N1. Мутации Y85E и N154K в HA2 являются критическими для размножения вируса в культуре клеток, а набор мутаций P215T в HA1 и W92G, D160H в HA2 дали вакцинному штамму существенное преимущество для размножения в культуре клеток MDCK, что может быть эффективно использовано в производстве культуральной ЖГВ.
Обсуждение. Изучение производственных серий культуральных ЖГВ показало наличие адаптационных мутаций в молекуле гемагглютинина штаммов H1N1 (K116E в субъединице НА2) и H3N2 (S219Y и N246K в субъединице НА1). Все изученные адаптационные мутации не влияли на антигенность вакцинных штаммов.
Заключение. Полученные в ходе исследования данные указывают на перспективность производства культуральной ЖГВ из реассортантных штаммов, подготовленных стандартным путём в РКЭ.
296-309
Встречаемость и генотипирование возбудителей клещевых инфекций в иксодовых клещах на востоке Западной Сибири (Россия, 2023 г.)
Аннотация
Введение. Томская область относится к регионам РФ с максимально высоким уровнем заболеваемости населения клещевыми инфекциями. Однако спектр и генетическое разнообразие возбудителей клещевых инфекций изучены недостаточно.
Цель исследования — оценить встречаемость и провести генотипирование различных видов возбудителей клещевых инфекций в иксодовых клещах, собранных с растительности в городских и пригородных биотопах г. Томска.
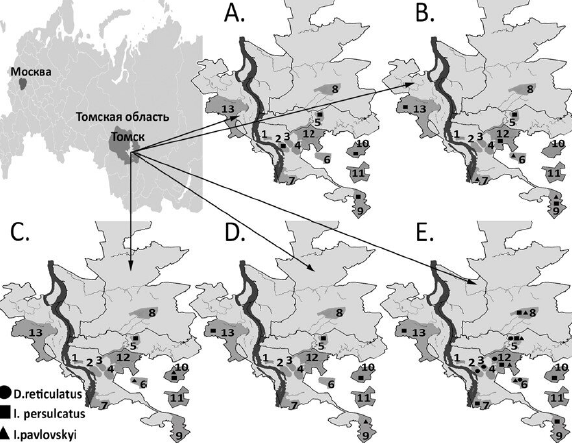
Материалы и методы. В исследовании проанализированы 534 клеща: Ixodes persulcatus (n = 107), I. pavlovskyi (n = 234) и Dermacentor reticulatus (n = 193), собранных в 13 биотопах Томска и в биотопах пригородов в течение 2023 г. Детекция генетического материала клещевых патогенов проведена методом полимеразной цепной реакции (ПЦР) и ПЦР с обратной транскрипцией в индивидуальных клещах с последующим секвенированием и филогенетическим анализом нуклеотидных последовательностей.
Результаты. Обнаружено более чем четырехкратное доминирование клещей I. pavlovskyi и D. reticulatus над таёжным клещом. При этом инфицированность клещей I. persulcatus вирусом клещевого энцефалита (ВКЭ) сибирского генотипа составила 1,3%, в клещах рода Ixodes генетический материал Borrelia burgdorferi s.l. был обнаружен в 8,5%, B. miyamotoi — 2,1%, Anaplasma phagocytophilum — 1,5%, а Rickettsia tarasevichiae — 14,1%. Инфицированность R. raoultii клещей D. reticulatus составила 48,7%, а в единичном образце была обнаружена ДНК Babesia canis. Генотипирование и филогенетический анализ геномных нуклеотидных последовательностей показал наличие новых, необычных для региона геновариантов B. garinii, B, bavariensis, B. afzelii и сибирского генотипа ВКЭ (субклайд V).
Заключение. На территории Томска и его пригородов в иксодовых клещах обнаружен генетический материал 9 видов клещевых патогенов, в том числе их новые генетические варианты.
310-324
Сочетанное действие гиперэкспрессии и мутаций гена ERG11 при формировании резистентности Candida albicans к триазоловым противогрибковым препаратам
Аннотация
Введение. Современная медицина сталкивается с резистентностью Candida spp. к антимикотикам, обусловленной изменением экспрессии и структуры гена ERG11 — молекулярной мишени триазолов. Эти механизмы часто действуют одновременно, однако взаимодействие между ними остаётся недостаточно изученным.
Цель работы — изучение роли гиперэкспрессии гена ERG11 и его мутаций в формировании резистентности грибов C. albicans к триазолам.
Материалы и методы. Исследование выполнено на 11 штаммах грибов C. albicans из коллекции МНИИЭМ им. Г.Н. Габричевского. Штаммы были охарактеризованы по уровню экспрессии гена ERG11 и наличию в нем мутаций, а также чувствительности к триазолам: позаконазолу, вориконазолу, итраконазолу и флуконазолу.
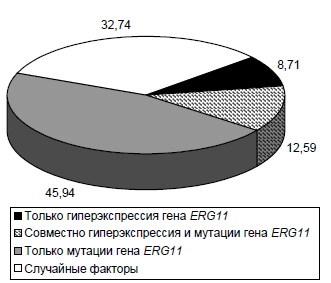
Результаты. Штаммы C. albicans подразделили на 4 группы: 1-я группа — только с повышенной экспрессией гена ERG11; 2-я — только с мутациями в данном гене; 3-я — одновременно оба вида генетических изменений; 4-я — без данных генетических изменений. Установлено, что минимальная подавляющая концентрация (МПК) триазолов в 1-й группе была в 15,76 раза выше, чем во 2-й, в 4,97 раза выше, чем в 3-й, и в 2,51 раза ниже, чем в 4-й (везде p < 0,05). Во 2-й группе МПК триазолов была в 3,17 раза ниже, чем в 3-й, и в 40 раз ниже (p < 0,001), чем в 4-й. МПК триазолов в 3-й группе по сравнению с 4-й группой была в 12,5 раза ниже (p < 0,001). Популяционное варьирование МПК триазолов в большей степени зависит от изолированного действия мутаций гена ERG11 (45,94%), что в 5,27 раза превосходит эффект изолированной гиперэкспрессии гена.
Заключение. Устойчивость C. albicans к триазолам обеспечивается кооперативным действием гиперэкспрессии и мутаций гена ERG11: наибольшую резистентность обеспечивает гиперэкспрессия, популяционное разнообразие — мутации.
325-330
Модели глобального прогнозирования вспышек денге в эндемичных регионах: систематический обзор
Аннотация
Введение. Лихорадка денге — быстро распространяющееся заболевание, переносимое комарами, представляет серьёзную проблему для глобального здравоохранения, особенно в эндемичных регионах. Частота и интенсивность вспышек лихорадки денге увеличиваются, что требует создания надёжных моделей прогнозирования для раннего вмешательства.
Цель систематического обзора — обобщить данные литературы о моделях прогнозирования лихорадки денге, оценить их прогностическую эффективность и выявить наиболее эффективные подходы.
Материалы и методы. Всесторонний поиск в базах данных Scopus, PubMed, ScienceDirect и Springer проведён в соответствии с рекомендациями PRISMA. Исследования отбирали на основе строгих критериев включения и исключения, а качество исследований оценивали с помощью критериев TRIPOD. Из 1366 выявленных исследований 13 соответствовали критериям отбора. Данные были проанализированы для оценки точности и обоснованности использованных моделей прогнозирования.
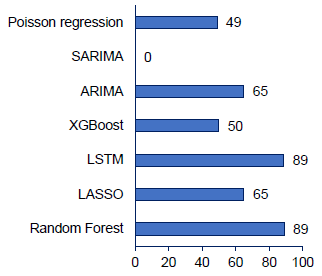
Результаты. Результаты показывают, что модели на основе машинного обучения, в частности «случайный лес», превосходят традиционные статистические модели, такие как ARIMA и регрессия Пуассона. Кроме того, климатические данные, особенно температура и количество осадков, играют важную роль в прогнозировании заболеваемости лихорадкой денге.
Заключение. Настоящее исследование подтверждает более высокую эффективность моделей прогнозирования на основе машинного обучения, в частности «случайный лес», в прогнозировании случаев заболевания лихорадкой денге по сравнению с традиционными статистическими методами. Этот вывод закладывает основу для разработки усовершенствованной системы раннего оповещения для борьбы с будущими вспышками лихорадки денге.
331-342
Полногеномное секвенирование двух клинических штаммов Mycobacterium tuberculosis c фенотипической чувствительностью к рифампицину при прогнозируемой Xpert MTB/RIF устойчивости
Аннотация
Введение. Более 40% штаммов Mycobacterium tuberculosis устойчивы к рифампицину (RIF) и изониазиду — препаратам первого ряда. Возбудитель туберкулёза приобретает устойчивость к RIF главным образом за счёт мутаций в гене rpoB.
Цель исследования — поиск наиболее вероятных компенсаторных мутаций в генах rpoA, rpoB и rpoC, кодирующих α-, β- и β′-субъединицы РНК-полимеразы M. tuberculosis.
Материалы и методы. Перекрёстный анализ фенотипической и генетической устойчивости к RIF среди 2298 клинических штаммов M. tuberculosis выявил 8 случаев, когда устойчивость, определённая тестом Xpert Ultra MTB/RIF, не подтверждалась бактериологическим методом. Во всех случаях это были хронические больные туберкулёзом с множественной или широкой лекарственной устойчивостью, у которых был отменён RIF по причине обнаружения устойчивости к этому препарату у выделенного штамма. Для исследования генотипа, секвенирования по Сэнгеру и полногеномного секвенирования были получены 2 штамма.
Результаты. Повторный тест Xpert Ultra MTB/RIF, секвенирование по Сэнгеру и полногеномное секвенирование выявили наличие единственной мутации S450L в гене rpoB при наличии фенотипической чувствительности у обоих штаммов. При филогенетическом анализе выяснено, что оба генома принадлежали к генотипу Beijing B0/W148. Штаммы отличались более высокой скоростью роста, чем другие изоляты. Выявлены две потенциальные компенсаторные мутации V483G и H748P в гене rpoС при отсутствии других значимых изменений в генах rpoA и rpoB.
Заключение. Высказано предположение, что феномен расхождения бактериологических и молекулярно-генетических результатов связан с приобретением в процессе лечения RIF штаммами Beijing B0/W148 компенсаторных мутаций в гене rpoС, а выявленные мутации влияют на конформацию β'-субъединицы, восстанавливая эффективность транскрипции, вызванную мажорной мутацией S450L.
343-349
Изучение патогенного потенциала и возможности межвидового перехода вирусов гриппа птиц подтипа Н5, выявленных на территории России в 2018–2022 годах
Аннотация
Введение. Высокая скорость эволюции вирусов высокопатогенного гриппа птиц (ВПГП), обусловленная антигенным дрейфом и реассортацией, может привести к устойчивой репликации и передаче вируса млекопитающим, что наблюдается в популяциях животных в последние годы. Исследование маркеров патогенности для млекопитающих у циркулирующих вирусов ВПГП даёт возможность оценить их патогенный потенциал и способность к межвидовому переходу.
Цель работы — провести анализ геномных последовательностей изолятов вируса гриппа птиц (ВГП) подтипа Н5, выявленных на территории России в 2018–2022 гг.
Материалы и методы. В работе использованы результаты собственного полногеномного секвенирования и нуклеотидные последовательности изолятов и штаммов ВГП подтипа Н5, опубликованные в открытых базах данных.
Результаты. Установлено, что преобладают вирусы с репликативным комплексом, адаптированным к размножению в клетках птиц. Анализ аминокислотной последовательности вирусного гемагглютинина выявил доминирование в рецептор-связывающем сайте белка аминокислот, характерных для ВГП и обеспечивающих повышенное сродство к рецепторам SAα-2,3-Gal эпителиальных клеток птиц. Показано появление и распространение в популяции ВГП факторов вирулентности для млекопитающих, таких как полноразмерный активный белок PB1-F2, дополнительная вставка из 5 аминокислот в белке NS1 и аминокислотные замены в белке M1.
Заключение. Наличие в популяции ВГП факторов патогенности для млекопитающих может способствовать успешному межвидовому переходу вируса за счёт подавления отдельных элементов иммунной защиты с последующей адаптацией вирусного гемагглютинина к клеточным рецепторам млекопитающих в результате антигенного дрейфа с дальнейшим закреплением приобретённых мутаций естественным отбором. Элиминация из популяции ВГП ряда адаптационных мутаций, способствующих размножению ВГП в клетках млекопитающих, подтверждает эффективность стратегии стемпинг аут и запрета на вакцинацию в промышленном птицеводстве в качестве сдерживающего фактора для гриппа птиц как зооантропонозного заболевания.
350-361
НАУКА И ПРАКТИКА
Усовершенствование бактериологического метода при выделении Listeria monocytogenes
Аннотация
Введение. Листериоз расценивается как одна из опасных вакцинонеуправляемых инфекций, характеризующаяся тяжестью клинического процесса и высокой летальностью. Актуальным направлением остаётся совершенствование методов лабораторной диагностики, особенно при листериозном менингите, для выявления возбудителя в оптимально сжатые сроки.
Цель исследования — изучить поведение музейных и клинических штаммов различных видов листерий на ГБМ-агаре — питательной среде для выделения возбудителей гнойных бактериальных менингитов — для усовершенствования бактериологического метода при выделении и идентификации Listeria monocytogenes.
Материалы и методы. В работе использованы 1125 образцов клинического материала и пищевых продуктов, 95 выделенных и 5 референтных штаммов Listeria spp., питательные среды для выделения листерий: агар Listeria пo Оттавиани и Агости (ALOA); питательный бульон для выделения и культивирования листерий (ПБЛ), питательный агар для выделения листерий (ПАЛ), ГБМ-агар.
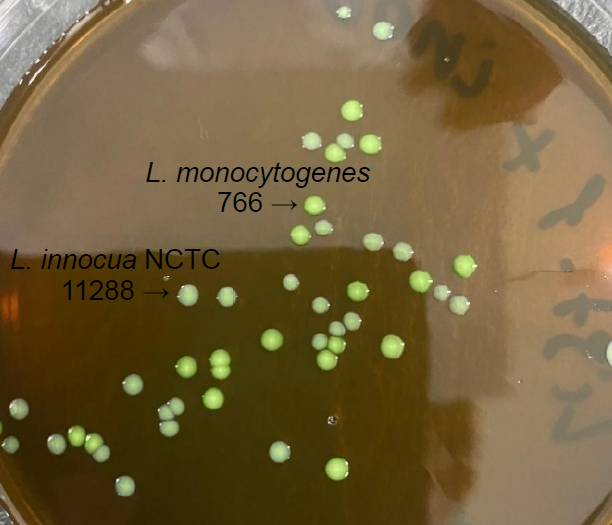
Результаты. Из 1125 образцов, поступивших на исследование, с использованием ПБЛ, ПАЛ и ALOA выделены штаммы: L. monocytogenes — 89, L. welshimeri — 2, L. innocua — 3, L. seeligeri — 1. Все изоляты и тест-штаммы субкультивировали на традиционные селективные среды (ПАЛ, ALOA) и дополнительно на ГБМ-агар, модифицированный внесением селективной добавки для выделения L. monocytogenes и желточной эмульсии. На модифицированном ГБМ-агаре выросшие колонии, относящиеся к роду Listeria, были крупнее и имели отличительные морфологические признаки от колоний, полученных на классических листериозных средах, что позволило провести первичную дифференциацию L. monocytogenes от непатогенных видов листерий и других возбудителей гнойных бактериальных менингитов.
Заключение. Показана возможность использования в алгоритме культурального метода новой питательной среды (модифицированный ГБМ-агар), обладающей улучшенными ростовыми свойствами в отношении L. monocytogenes, внедрение которой послужит дополнительным эффективным средством для дифференциации листерий при проведении исследований в санитарной и клинической микробиологии.
362-369
ОБЗОРЫ
Органоидные (3D-клеточные) культуры в оценке способности вирусов к межвидовым переходам

Аннотация
Цель обзора — охарактеризовать возможности применения органоидных (3D-клеточных) культур для оценки способности вирусов к межвидовым переходам.
Использованы источники из баз данных Web of Science, PubMed, Scopus, Elsevier, Google Scholar и eLIBRARY.RU по состоянию на февраль 2025 г.
В работе системы эпидемиологического надзора, помимо классических методов эпидемиологической диагностики и надзора за вирусными инфекциями, широко применяются молекулярно-генетические технологии (полимеразная цепная реакция и секвенирование). Как показывает передовой мировой опыт, в решении этих вопросов перспективным является использование органоидных (3D-клеточных) культур. В настоящем обзоре проанализированы данные по применению органоидных (3D-клеточных) культур человеческого и животного происхождения для изучения иммунопатогенеза, а также оценки способности ряда вирусов (SARS-CoV-2, гриппа, Зика, кори и др.) к межвидовым переходам, что обусловливает их пандемический потенциал.
370-380
ЮБИЛЕИ
К 60-летию академика РАН Василия Геннадьевича Акимкина
 381-382
381-382
К 100-летию профессора Натальи Николаевны Костюковой
 383-383
383-383
НЕКРОЛОГИ
Памяти Виталия Александровича Романова
 384-384
384-384
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.