


Phylogeny of Yersinia pestis strains of the 4.ANT lineage from the Tuva mountains and adjacent plague foci
- Authors: Stankovtseva E.V.1, Oglodin E.G.1, Verzhutsky D.B.2, Chervyakova N.S.1, Naryshkina E.A.1, Fedorov A.V.1, Eroshenko G.A.1, Balakhonov S.V.2, Kutyrev V.V.1
-
Affiliations:
- Russian Anti-Plague Institute «Microbe»
- Irkutsk Research Anti-Plague Institute of Siberia and Far East
- Issue: Vol 102, No 3 (2025)
- Pages: 284-295
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18773
- DOI: https://doi.org/10.36233/0372-9311-659
- EDN: https://elibrary.ru/SCLRIW
- ID: 18773

Cite item

Abstract
Introduction. The Tuva mountain plague focus (TMPF) in Russia has been continuously epizootically active since its discovery in 1964. The strains of Yersinia pestis isolated in this focus belong to the phylogenetic lineage 4.ANT of the antique biovar of the main subspecies. They are highly virulent and epidemically significant. The use of modern molecular genetic technologies will make it possible to determine the population structure of 4.ANT strains in the TMPF.
The aim of the study was to analyze the phylogenetic and population structure of Y. pestis strains of the 4.ANT lineage from the TMPF according to the data of whole-genome SNP (single nucleotide polymorphism) typing and MLVA25 (multiple locus variable number tandem repeats analysis) typing.
Materials and methods. Whole-genome nucleotide sequences of 68 Y. pestis strains, including 60 strains of the 4.ANT lineage, were analyzed. Sequencing of strains was performed on the MGI platform. SNP-analysis was performed by sequence alignment in the Snippy v. 4.6 program with subsequent construction of a Maximum Likelihood dendrogram based on the identified core SNPs in the SeaView program. SNPs, being markers for strains of the 4.ANT lineage, were detected using the MEGA11 program. MLVA-genotyping of Y. pestis strains of the 4.ANT lineage was performed by searching loci and then counting the number of tandem repeats in the Tandem Repeats Finder program. MLVA-dendrogram construction was performed by UPGMA method in the BioNumerics v. 7.6.3 program.
Results. According to SNP-analysis of Y. pestis strains of lineage 4.ANT from the TMPF, the presence of 4 phylogeographic groups was established: T1 (Saglinsky, Tolaylyg and Barlyk mesofoci, 1971–1987), T2 (Karginsky mesofocus, 2014–2024), T3 (Karginsky mesofocus, 1977–2009), T4 (Karginsky, Tolaylyg and Boro-Shai mesofoci, 2006–2013). Eight MLVA-genotypes of strains of 4.ANT lineage from Tuva and variable VNTR loci were identified: yp1290ms04, yp1935ms05, yp0559ms15, yp4042ms35, yp4425ms38, yp1108ms45, yp4280ms62, yp1580ms70.
Discussion. Among the strains analyzed, the earliest representatives of the 4.ANT branch are strains of the T1 cluster from the TMPF. The population of strains from the Altai Mountains and Mongolia and the population of strains from the TMPF (1977–2024) are represented as separate sub-branches on the tree. The latter population is represented by polytomy and is characterized by pronounced clustering according to the spatial and temporal principle.
Conclusion. The presence of 4 main phylogeographic groups in the population of 4.ANT lineage in the TMPF was determined and genetic differences between them were established, which can be used for in-depth molecular-genetic differentiation and typing of Y. pestis strains in this focus.
Keywords
Full Text
Introduction
Natural foci of plague are located on most continents and are constantly manifested by outbreaks of this particularly dangerous infection, which has left a deep trace in the history of civilization in the prehistoric period as well as the modern era [1]. In recent years, outbreaks of plague have been reported in the Democratic Republic of the Congo, the Republic of Madagascar, the United States of America, the People's Republic of China and Mongolia [2]. Plague is a natural focal infection with a predominantly vector-borne mechanism of transmission of the pathogen, the bacterium Yersinia pestis, which persists in natural foci, mainly circulating between rodents and fleas parasitizing them. There are 45 natural foci of plague in the countries of the Commonwealth of Independent States, including 11 of them in Russia.
The modern intraspecific classification, based on data on the global genetic diversity of the plague pathogen, divides Y. pestis strains into 7 subspecies: the major subspecies — ssp. pestis (ancient, medieval, eastern and intermediate biovars) and 6 non-major subspecies [3]. Strains of the major subspecies circulate in most natural foci of the world and are highly virulent. Strains of the ancient biovar were etiologic agents of the 1st and 2nd plague pandemics, the Manchurian epidemic of pneumonic plague in China in 1910–1911, and modern outbreaks of plague in the Democratic Republic of Congo [3, 4]. Antique biovar strains are genetically diverse and belong to 5 phylogenetic lineages according to the genetic nomenclature of evolutionary branches: 0.ANT, 1.ANT, 2.ANT, 3.ANT and 4.ANT, which are currently found in natural plague foci in Asia and Africa [5]. Strains of the 4.ANT lineage circulate in an endemic megafocus, which is transboundary and covers the territories of the TMPF and the Gorno-Altaisk high-mountain plague focus in Russia and natural foci in Mongolia [6]. 4.ANT strains are not found in other regions of the world. For many years, the 4.ANT mega-focus has shown constant epizootic activity. In 2014–2016, 3 cases of human plague caused by strains of the 4.ANT lineage occurred in the Gorno-Altaisk highland focus [7, 8]. Plague cases are also registered in the neighboring region of Mongolia [2, 9].
In addition to the 3 virulence plasmids pFra, pCad, and pPst resident to Y. pestis, 4.ANT strains contain the pTP33 plasmid, which apparently encodes adaptation factors of Y. pestis strains to the conditions of natural ecosystems in this geographic region [10, 11]. 4.ANT strains have been isolated in the Gorno-Altaisk focus since 2012. The phylogenetic structure of 4.ANT strains from the Altai Mountains and Mongolia has been well investigated using whole genome sequencing and MLVA25-typing [6, 12]. Circulation of the Tuva variant of 4.ANT in the TMPF was detected as early as 1964. [13]. Since then, epizootic activity in the TMPF has been recorded continuously and with the isolation of Y. pestis cultures, but the number of publications on molecular genetic studies of the population structure of Tuvan strains of 4.ANT is rather limited [6, 12, 14, 15]. There are practically no publications on phylogenetic analysis of the population structure of 4.ANT strains based on the data of whole genome sequencing.
The TMPF covers 3 administrative districts of Tuva: Mongun-Taiginsky, Ovyursky and Tes-Khemsky. The main territory of the focus is located near the southern slopes of the Tsagan-Shibetu and Western Tannu-Ola mountain ranges [16]. The territory of the focus includes various geographical landscapes: from the steppe zone to alpine biotopes. The main feature of the epizootic process in the focus is a pronounced microfoci, which is directly related to the presence of separate populations of the main carrier — the long-tailed gopher Urocitellus undulatus. The TMPF includes a number of mesofoci: Karginsky, Saglinsky, Tolaylygsky, Barlyksky, Verkhne-Barlyksky, Boro-Shaysky, Mogen-Burensky, Aspaitinsky, Kara-Beldyrsky, Chozinsky and Despensky [17]. The main vector in the territory of the focus is the flea Citellophilus tesquorum, however, other species of fleas, ixodes and gamaze ticks, and lice are also involved in the epizootic process [18]. The existence of separate plague mesofoci and micro-foci of the territory implies the presence of different phylogeographic populations and a pronounced diversification of the 4.ANT lineage in the TMPF. The combination of the methods of whole-genome SNP analysis (single nucleotide polymorphism) and MLVA25 typing (multiple locus variable number tandem repeats analysis) has proven to be an effective genetic tool for determining the population structure of Y. pestis [19, 20]. The first method allows reconstruction of the long-term evolution of Y. pestis, and MLVA25 shows high resolution when studying closely related strains circulating in the same or adjacent territories [21, 22].
The TMPF is one of the active plague foci of Russia. The southern part of the focus is adjacent to the border with Mongolia, where active plague foci are located. The development of tourism, economic ties and transportation in this region may lead to cases of human plague infection and transfer of the pathogen outside the epizootic areas. The planned construction of the Elegest — Kyzyl — Kuragino railroad in 2026 may also increase the threat of contact with carriers and vectors of the disease. Another threat is the illegal harvesting of the marmot tarbagan by local people, which has been sporadically involved in epizootics in the last 10–15 years. The intensity of the epizootic process in the TMPF and the high virulence of 4.ANT strains necessitate their comprehensive study, determination of their range, phylogenetic analysis and establishment of the current population structure using molecular genetic technologies.
The aim of this study is the phylogenetic and population analysis of Y. pestis strains of lineage 4.ANT from TMPF according to whole genomic SNP- and MLVA25-typing data.
Materials and methods
Whole-genome SNP analysis of Y. pestis strains of the 4.ANT phylogenetic lineage
The whole-genome nucleotide sequences of 68 Y. pestis strains were used in this study, of which 60 strains isolated in 1971–2024 belong to the phylogenetic lineage 4.ANT. Among them, 53 strains were obtained from the TMPF, 5 strains from the Gorno-Altaisk focus and 2 strains from Mongolia. Strains from the TMPF were isolated from the long-tailed gopher Urocitellus undulatus (26%), tarbagan Marmota sibirica (4%); Daurian pika Ochotona dauurica (4%), lice (11%), fleas Citellophilus tesquorum (35%), Oropsylla alaskensis (4%), Paramonopsyllus scallonae (2%), Rhadinopsylla li transbaikalica (6%), Frontopsylla elatoides (4%); from Gamasina (2%), Ixodidae (2%) ticks. Y. pestis strains were obtained from the State Collection of Pathogenic Bacteria of the Russian Anti-Plague Institute “Microbe” of Rospotrebnadzor.
Strains were grown on agar or LB broth (pH 7.2) at 28℃ for 24–48 hours. DNA was isolated using the PureLink Genomic DNA Mini Kit (Invitrogen) according to the manufacturer's instructions. Nucleotide sequences of Y. pestis strains were obtained by whole genome sequencing on MGI platform (DNBSEQ-G50RS sequencer) using MGIEasy Fast FS Library Prep Set and MGIEasy UDB Primers Adapter Kit A reagent kits. The resulting reads (DNA fragments produced by the sequencer) were assembled into contigs (a set of overlapping DNA segments that together represent the consensus DNA region) with an average coverage per genome of 98.56% (50× read depth). The average size of the assembled genome was 4.55 million nucleotide pairs. Y. pestis strains of different phylogenetic lineages from the NCBI GenBank database were taken as a comparison group for dendrogram construction: 620024 (NZ_ADPM000000000000. 1, 0.PE7), Pestoides A (NZ_ACNT000000000000.1, 0.PE4), Antiqua (NC_008150.1, 1.ANT), CO92 (NC_003143.1, 1.ORI1), KIM10 (NC_004088. 1, 2.MED1), Nepal516 (NC_008149.1, 2.ANT1), MGJZ11 (NZ_ADSU000000000000.1, 3.ANT2), MGJZ12 (NZ_ADSV000000000000.1, 4.ANT). Sequences of certain 4.ANT strains were also taken from the NCBI GenBank database: I-3113 (NZ_CP045149.1, 4.ANT), I-3223 (LZNE00000000.1, 4.ANT), 131–133 (M2085) (NZ_CP064125.2, 4.ANT), 256 (M2029) (NZ_CP064123.1, 4.ANT).
Core SNP mutations were detected by aligning Y. pestis strain contigs to the Y. pestis CO92 genome using the Snippy v. 4.6 program, then 28 homoplasy SNPs were removed, which appear independently in representatives of different phylogenetic lineages and do not reflect the unity of origin [5]. The resulting file contained 1133 core SNPs. The PhyML module in the SeaView program was used to construct a dendrogram based on core SNPs. Maximum Likelihood dendrogram with nucleotide substitution model — GTR (general time reversible) was visualized in the FigTree v. 1.4.5 program. The search for marker SNPs was performed in the MEGA11 program.
MLVA25-genotyping of Y. pestis strains of the 4.ANT phylogenetic lineage
Genotyping was performed at 25 VNTR loci with exclusion of the yp3057ms09 locus from the analysis [23, 24]. VNTR loci were searched using the FragmentFinder v. 0.4 program [25]. The number of tandem repeats was counted in the Tandem Repeats Finder program under the following parameters: alignment parameters — 2, 3, 5 (match, mismatch, indel, respectively); minimum match score to report the presence of a repeat — 50; maximum period size (the program's best guess for the size of the tandem repeat template) — 500 bp [26]. A dendrogram based on the number of tandem repeats was constructed in the BioNumerics v. 7.6.3 program (Applied Maths) using the UPGMA method (unweighted pair group method with arithmetic mean).
Statistical data processing included calculation of the allele polymorphism index h and evaluation of the discriminatory ability of the method by calculating the Hunter-Gaston index [27, 28].
Results
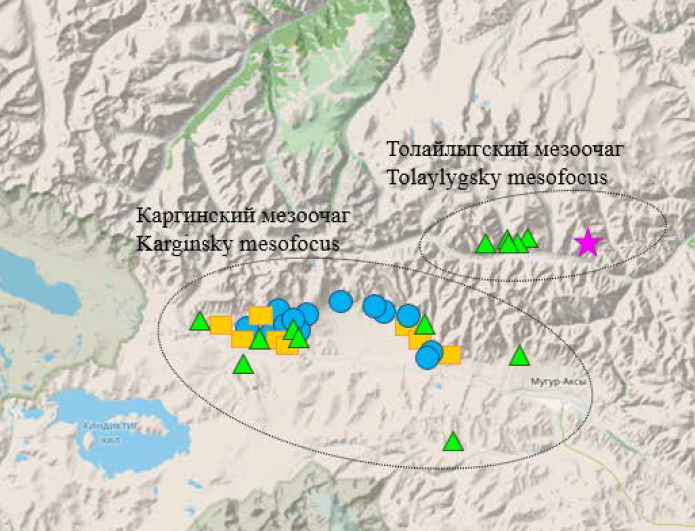
Based on the results of whole-genome SNP analysis, a phylogenetic study was performed and the population structure of Y. pestis strains of the 4.ANT lineage from the TMPF was determined, which was found to include 4 major phylogeographic groups (Fig. 1). The phylogenetic relatedness of the strains was reconstructed on the basis of 1133 identified bark SNPs. The phylogenetic tree in Fig. 1 is rooted using Y. pestis strain 620024 (NCBI GenBank: NZ_ADPM000000000000.1, 0.PE7) [5]. The locations of isolation of Y. pestis strains of line 4.ANT in TMPF are shown in Fig. 2.

Fig. 1. Maximum Likelihood dendrogram of Y. pestis strains of the 4.ANT phylogenetic lineage, constructed from the data of whole genome SNP analysis based on 1133 bark SNPs.
The PhyML module of the SeaView program was used to construct the dendrogram. The model of nucleotide substitutions — GTR with 500-fold bootstrap support was used. FigTree v. 1.4.5 program was used for dendrogram visualization. To improve the resolution of the figure, the branch of strain 620024 is not shown in the dendrogram.
mesof. — mesofocus, n. l. — natural landmark (tract).

Fig. 2. Spread of Y. pestis strains of phylogroups T1–T4 of the 4.ANT lineage in the territory of the TMPF.
The search for SNP mutations underlying the separation of strains of the 4.ANT lineage from the common stem of the phylogenetic tree of Y. pestis revealed 12 specific SNPs common to all strains of the 4.ANT lineage. Of these, 9 SNPs are located in genes encoding cell life support proteins, including 6 nonsynonymous SNP mutations. Another 3 SNP mutations are located in the intergenic space. One of the identified SNP mutations with coordinate 1610851 in the genome of Y. pestis strain CO92 (G→A, rlmKL gene) was previously used as a target for detection of strains of the 4.ANT lineage in allele-specific qPCR [29].
In the dendrogram, Y. pestis strains of the 4.ANT lineage isolated in the TMPF area separated into four phylogenetic clusters (phylogroups). Cluster T1, which separated from the trunk of the 4.ANT lineage earlier than the others, included 6 strains obtained in 1971–1987. These are some of the earliest strains from the TMPF in the study sample. Strains 2060, 1771, I-3110 were obtained in the Sagli mesofocus in 1971 and 1984. Strain I-3205 (1986) was isolated in the Tolaylyg mesofocus (Buure tract). The genome of strain I-3113 (1984) was taken from the NCBI GenBank database (NZ_CP045149.1). Strains I-3113 and I-3223 (1987) were isolated in the Barlyk mesofocus. Fourteen SNP mutations characteristic only of strains of the T1 cluster were detected, of which 12 SNP mutations were located in the coding region (9 nonsynonymous), and 2 mutations were located in the intergenic space. The branch that gave rise to the remaining strains of the 4.ANT lineage branches off from the phylogenetic node common to the T1 cluster.
Between the strains of cluster T1 and other strains of the 4.ANT lineage in the dendrogram is strain MGJZ12, which is a member of the comparison group strains [5]. This strain was isolated in Mongolia in 2002, which indicates phylogenetic continuity of 4.ANT strains distributed in this transboundary area of natural plague foci.
A separate cluster on the dendrogram is formed by strains from the Altai Mountains and Mongolia. Strain I-3240 was isolated earlier than other strains of this cluster (1988) in the territory of the Khukh-Serkh-Munkh-Hairkhan focus (Mongolia). Modern strains from the Mountain Altai and Mongolia (2012–2019) originate from a common ancestor with strain I-3240. These strains belong to a new powerful clone of the 4.ANT lineage, which manifested itself in the second decade of the 21st century in the transboundary section of the 4.ANT megafocus, including cases of human plague in Russia and Mongolia [7, 8]. Four specific SNP mutations were identified for strains from the Gorno-Altaisk highland focus of Russia and Mongolia, located in coding regions of the genome.
All other strains from the TMPF taken in the study, represented by a separate branch on the dendrogram, lie at the base of the overall polytomy. This branch includes three different clusters, designated as T2-T4, as well as individual strains (I-3462, 2002; I-3471, 2003; I-3383, 1994; I-3401, 1998) that did not fall into any cluster. All of these strains were isolated in the Kargi mesofocus.
Cluster T2 included 10 strains isolated in 2014-2024 in the Karginsky mesofocus. They are separated from other clusters by the presence of 2 specific SNP mutations, one of which is located in the coding region with coordinate 2774153 on the CO92 genome (G→A, gene YPO_RS13360, synonymous).
Cluster T3 includes strains obtained in the Karginsky plague mesofocus in 1977–2009. This large cluster is formed by strains with a similar SNP profile. Diversification of individual subclusters within cluster T3 in combination with isolation of cultures of the plague pathogen during 40 years in the territory of this mesofocus suggests the presence of an independent process of microevolution of strains of cluster T3 during that period. The strains of the T3 cluster have one common specific SNP mutation with the coordinate 4263645 on the CO92 genome (G→T, gene YPO_RS20065, nonsynonymous).
4.ANT strains of another cluster, T4, were isolated in the Tolaylyg and Karginsky mesofoci of the TMPF in 2006-2013. Strain 3 (2012), isolated in the Boro-Shai mesofocus, was also included in the T4 cluster. Three SNP mutations specific for strains of this cluster were detected, 2 SNP mutations are nonsynonymous and located in the coding region (325289, T→A, gene YPO_RS02605; 3972331, C→T, gene YPO_RS18755).
Thus, the presence of diversification of Y. pestis 4.ANT strains in the TMPF due to independent microevolution of the pathogen in isolated plague microfoci was established on the basis of a whole-genome SNP analysis, and the main phylogeographic groups of these strains were described. SNP mutations specific for individual phylogeographic populations of this line of evolution of the plague pathogen are characterized.
MLVA25-genotyping of Y. pestis strains of the 4.ANT phylogenetic lineage from the TMPF
MLVA25-genotyping was carried out for all 60 Y. pestis strains of the 4.ANT lineage obtained in 1971–2024 in the TMPF, the Gorno-Altaisk high-mountain focus in Russia, and foci of Mongolia. Based on the typing results, 11 MLVA-genotypes were identified (Hunter-Gaston index equal to 0.78). The following loci were variable for 4.ANT strains: yp1290ms04 (number of tandem repeats 6, 7); yp1935ms05 (4, 9); yp0559ms15 (8, 9); yp4042ms35 (9, 10); yp4425ms38 (5, 8); yp1108ms45 (6, 7); yp3060ms56 (8, 9); yp4280ms62 (6, 7, 9, 10, 11, 12, 13, 14); yp1580ms70 (4, 5, 6) (Table). For the remaining loci (yp0120ms01; yp2769ms06; yp2916ms07; yp1814ms20; yp1895ms21; yp0581ms40; yp0718ms41; yp1018ms44; yp1335ms46; yp2058ms51; yp2612ms54; yp1118ms69; yp1925ms71; yp3236ms73; yp3245ms74), all strains appeared identical. Strains from TMPF formed 8 MLVA genotypes. The same loci were found to be variable for them as for all strains of the 4.ANT lineage, with the exception of yp3060ms56 (8).
Characterization of variable VNTR loci of Y. pestis strains of the 4.ANT lineage by MLVA25 genotyping
VNTR locus | Repeat length, bp | Number of alleles and repeat copies in the VNTR locus | Allelic polymorphism index h |
yp1290ms04 | 17 | 6, 7 | 0,19 |
yp1935ms05 | 17 | 4, 9 | 0,03 |
yp0559ms15 | 15 | 8, 9 | 0,35 |
yp4042ms35 | 15 | 9, 10 | 0,19 |
yp4425ms38 | 16 | 5, 8 | 0,10 |
yp1108ms45 | 12 | 6, 7 | 0,03 |
yp3060ms56 | 16 | 8, 9 | 0,21 |
yp4280ms62 | 9 | 6, 7, 9, 10, 11, 12, 13 | 0,74 |
yp1580ms70 | 9 | 4, 5, 6 | 0,44 |
When constructing the MLVA25-dendrogram using the UPGMA method based on the number of tandem repeats in the VNTR loci, all the strains from TMPF were divided into 3 major clusters: A and B (Fig. 3). The division of strains into clusters and subclusters coincides with their spatial and temporal origin in the focus: cluster A consists of strains from 1971–1987 from the Saglinsky, Tolaylygsky and Barlyk mesofoci; cluster B is formed by strains from 1977–2024. Cluster B includes subclusters: B1 — territory of Karginsky mesofocus (Chalyyash and Kok-Dorgun tract); B2 — Tolaylyg and Karginsky mesofoci; B3 — Karginsky mesofocus; B4 — Karginsky and Boro-Shai mesofoci. A separate cluster on the dendrogram is formed by strains from the Altai Mountains and Mongolia.

Fig. 3. MLVA-dendrogram of Y. pestis strains of the 4.ANT phylogenetic lineage obtained in 1971–2024 in the TMPF, the Gorno-Altaisky high-mountain focus in Russia and foci of Mongolia according to MLVA25-genotyping data, constructed by the UPGMA method.
mesof. — mesofocus, n. l. — natural landmark (tract)
Cluster A, as in the whole-genome SNP analysis, included some of the most previously isolated strains from Tuva. These are 3 strains from the Sagli mesofocus (I-3110, 1984; 2060 and 1771, 1971), strain I-3205 from the Tolaylyg mesofocus (1986), and strains from the Barlyk mesofocus (I-3223, 1987; I-3113, 1984). The MLVA profile of this group is very different from the other strains of the 4.ANT lineage. The presence of 2 alleles at the yp4425ms38 locus (5 and 8) and 2 alleles at the yp1108ms45 locus (6 and 7) underlies the formation of three MLVA genotypes in the strains of cluster A: Tuv.6, Tuv.7 and Tuv.8. Strains I-3110, 2060 and 1771 from the Sagli mesofocus have 8 tandem repeats in the VNTR yp4425ms38 locus and 6 repeats in the yp1108ms45 locus (genotype Tuv.6). Strains I-3205 (1986) and I-3223 (1987) have the Tuv.7 genotype, characterized by the presence of 5 repeats at the yp4425ms38 locus and 6 repeats at the yp1108ms45 locus. Strain I-3113 (1984), which has 5 repeats at the yp4425ms38 locus and 7 tandem repeats at the VNTR yp1108ms45 locus, belongs to a separate genotype, Tuv.8.
Cluster B was formed by all other studied Y. pestis 4.ANT strains from the 1977–2024 TMPF isolation. This is a fairly homogeneous group in terms of MLVA profile. Only the presence of 3 alleles at the yp4280ms62 locus (11, 12, 13) and 2 alleles at the yp1580ms70 locus (4, 5) underlies the division of the strains into subclusters B1, B2, B3, and B4 as well as the formation of 4 genotypes (Tuv.4, Tuv.1, Tuv.2, Tuv.3), respectively.
Subcluster B1 was formed by 2 strains from the Karginsky mesofocus — I-2638 (1977, Kok-Dorgun tract) and I-3403 (1998, Chalyyash tract). The formation of the Tuv.4 genotype is based on the presence of 4 repeats in the yp1580ms70 locus. The most similar MLVA-profile has strains of cluster B3, which on the tree originate from strains of cluster B1. Subcluster B3 includes strains isolated in 1992–2015 in the Kargin mesofocus. Cluster B1 and B3 strains (genotypes Tuv.4 and Tuv.2) are united by the presence of 12 tandem repeats at the yp4280ms62 locus. Subcluster B2 (genotype Tuv.1) is formed by 18 strains isolated in the Tolaylyg and Karginsky mesofoci (2002–2013). The strains of genotype Tuv.1 have 11 VNTR repeats at the yp4280ms62 locus. The presence of 13 repeats at the yp4280ms62 locus separates the 7 strains that formed subcluster B4 (2002–2024). This included strains from the Karga mesofocus, as well as strain 3 (2012) from the Boro-Shai mesofocus. The Y. pestis strain 549, isolated in 2020 in Kyzyl-Bom tract (Karginsky mesofocus), was not included in any cluster formed by other strains from the TMPF. This is the only strain that has 5 tandem repeats at the yp1935ms05 locus, which accounts for its characteristic MLVA-genotype Tuv.5.
A separate cluster on the dendrogram was formed by strains from the Altai Mountains and Mongolia (genotypes Alt.1, Alt.2, Mon.1).
Discussion
In Tuva, the Altai Mountains and the adjacent territory of Mongolia there is a natural megafocus of plague, in which strains of Y. pestis of the phylogenetic lineage 4.ANT of the antique biovar of the main subspecies endemic to this region are distributed. They are highly virulent and epidemically significant. The use of modern molecular genetic technologies is necessary to analyze the population structure and directions of microevolution of 4.ANT strains, to determine the diversity of genotypes and areas of their distribution, which is important for improving the efficiency of epidemiological monitoring in these active foci of Siberia. Over the last few years, the use of whole-genome SNP analysis and MLVA typing has proved the effectiveness of these methods for tracking the evolution and typing of Y. pestis strains, as well as in epidemic investigations and for controlling plague epizootics [6, 12, 15, 20–23, 30, 31].
Our phylogenetic study of 60 strains of Y. pestis lineage 4.ANT from the natural megafocus of plague, based on the data of whole-genome SNP-analysis, showed that the strains from the 1971–1987 TMPF were the earliest to diverge from the evolutionary trunk of this lineage. On the phylogenetic tree, strains from this period formed a closely related cluster, which included strains isolated in the Sagli (1971, 1984), Barlyk (1984, 1987), and Tolaylyg (1986) mesofoci. The strains were first isolated in this area in the Saglinsky mesofocus in 1966, in the Barlyksky mesofocus in 1983, and in the Tolaylygsky mesofocus in 1985 [16]. After large-scale disinfestation activities in 1981–1985, Y. pestis cultures were no longer isolated in the Saglinsky mesofocus. The performed phylogenetic analysis showed that the Tuvan strains from the 1971–1987 cluster are evolutionarily earlier and precede on the dendrogram all other strains of Y. pestis from the 4.ANT megafocus in Tuva and the Altai Mountains.
The strains of this cluster are followed on the dendrogram by two modern branches of evolution, one of which includes 4.ANT strains from 1988–2019 from the Gorno-Altaisk high-mountain focus in Russia and foci in Mongolia (Sailugemsky and Khukh-Serkh-Munkh-Hairkhansky). The second branch of 4.ANT consists of Tuvan strains from 1977–2024 predominantly from the Karginsky mesofocus. The SNP profile of this branch of Tuvan strains differs significantly from the strains of the 1971–1987 cluster, suggesting a subsequent independent microevolution of 4.ANT in the Kargin mesofocus. This branch of Tuvinian strains shows spatial and temporal clustering and their diversification within separate clusters, which indicates the ongoing process of independent microevolution of the 4.ANT lineage in the TMPF.
It was previously shown that the MLVA25-typing method has a significant discriminatory ability with respect to strains of Y. pestis of the major and non-major subspecies from the TMPF and the Gorno-Altaisk high-mountain plague focus, respectively [15]. It was shown that strains were clustered on the basis of the number of tandem repeats both at the population level (separation of strains depending on the focus) and at the intrapopulation level (separation of strains within one focus). Our data confirm the diversity of MLVA25-genotypes of 4.ANT strains isolated in the Tyva Republic, Gorny Altai and Mongolia. The data of MLVA25- and SNP-typing coincide, which proves the prospect of integrated use of these two modern methods to reconstruct the long-term evolution and analyze the population structure of 4.ANT strains. High discriminatory capabilities of the MLVA25 method in determining the intrapopulation structure of Y. pestis strains will allow further effective monitoring of the genetic variability of the plague pathogen in the natural megafocus of 4.ANT in Tuva and the Altai Mountains.
Conclusion
The phylogenetic structure of 4.ANT strains from the plague megafocus located in Russia and Mongolia was determined based on the data of whole-genome SNP analysis of 60 Y. pestis strains of the 4.ANT lineage from the plague megafocus, reflecting the spatial and temporal circulation of the pathogen in the megafocus. The presence of 4 major phylogeographic groups of 4.ANT strains from the TMPF was established. Phylogroup T1 includes strains from the Sagli, Barlyk, and Tolaylyg mesofoci of 1971–1987. Phylogroup T2 includes 10 strains isolated from 2014–2024 in the Karginsky mesofocus. Phylogroup T3 includes strains from the Karginsky mesofocus obtained in 1977–2009. Phylogroup T4 consists of strains isolated in 2006–2013 from Karginsky, Tolaylygsky and Boro-Shai mesofoci. Marker SNP-mutation dendrograms for phylogenetic nodes of 4.ANT were identified, which can be used for extended molecular genetic identification of strains from the TMPF. Using MLVA25-typing, the presence of 8 MLVA-genotypes for the Tuvan population of 4.ANT was established and variable VNTR loci were identified. The revealed genetic diversity of Y. pestis strains of the 4.ANT lineage is associated with microevolution of separate phylogeographic groups in different microfoci in the TMPF. The pronounced diversification distinguishes the 4.ANT population from the TMPF from the 4.ANT population from the Gorno-Altaisk focus, which is characterized by significant genetic homogeneity.
Thus, strains of lineage 4.ANT from the transboundary plague megafocus in Russia and Mongolia are a convenient model for studying the influence of existence conditions on the microevolution of different phylogeographic populations of Y. pestis. The obtained results of whole-genome SNP-analysis and MLVA25-typing can be used for molecular-genetic differentiation of Y. pestis strains of lineage 4.ANT from the TMPF, detailing the molecular-genetic passportization of the territory and increasing the efficiency of molecular-epidemiological monitoring of the TMPF and adjacent plague foci of Russia and Mongolia. Against the background of the growing tourist flow and construction of new transportation networks, the obtained data may contribute to reducing the risks of human plague as well as carrying the pathogen outside the epizootic territories.
About the authors
Elizaveta V. Stankovtseva
Russian Anti-Plague Institute «Microbe»
Author for correspondence.
Email: stankovtseva2101@yandex.ru
ORCID iD: 0009-0000-4735-3311
junior researcher, Laboratory of molecular microbiology
Russian Federation, SaratovEugeniy G. Oglodin
Russian Anti-Plague Institute «Microbe»
Email: e.oglodin@rambler.ru
ORCID iD: 0000-0002-2955-3034
Cand. Sci. (Biol.), leading researcher, Laboratory of Molecular microbiology
Russian Federation, SaratovDmitry B. Verzhutsky
Irkutsk Research Anti-Plague Institute of Siberia and Far East
Email: verzh58@rambler.ru
ORCID iD: 0000-0002-5139-616X
D. Sci. (Biol.), сhief researcher, Zoological and parasitology department
Russian Federation, IrkutskNadezhda S. Chervyakova
Russian Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0003-3133-3820
Cand. Sci. (Biol.), senior researcher, Department of "State Collection of Pathogenic Bacteria"
Russian Federation, SaratovEkaterina A. Naryshkina
Russian Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0002-9190-099X
researcher, Laboratory of genomic and proteomic analysis
Russian Federation, SaratovAndrey V. Fedorov
Russian Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0001-7190-4427
junior researcher, Laboratory of genomic and proteomic analysis
Russian Federation, SaratovGalina A. Eroshenko
Russian Anti-Plague Institute «Microbe»
Email: geroshenko@yandex.ru
ORCID iD: 0000-0001-5403-989X
D. Sci. (Biol.), сhief researcher, Laboratory of molecular microbiology
Russian Federation, SaratovSergey V. Balakhonov
Irkutsk Research Anti-Plague Institute of Siberia and Far East
Email: adm@chumin.irkutsk.ru
ORCID iD: 0000-0003-4201-5828
D. Sci. (Med.), Professor, Director
Russian Federation, IrkutskVladimir V. Kutyrev
Russian Anti-Plague Institute «Microbe»
Email: rusrapi@microbe.ru
ORCID iD: 0000-0003-3788-3452
D. Sci. (Med.), Professor, Academician of the RAS, Director
Russian Federation, SaratovReferences
- Попов А.Ю., Кутырев В.В. Атлас природных очагов чумы России и зарубежных государств. Саратов;2022. Popov A.Yu., Kutyrev V.V. Atlas of Natural Plague foci in Russia and Foreign Countries. Saratov;2022.
- Попов Н.В., Карнаухов И.Г, Кузнецов А.А и др. Эпидемиологическая ситуация по чуме в мире. Прогноз эпизоотической активности природных очагов чумы Российской Федерации на 2024 г. Проблемы особо опасных инфекций. 2024;(1):67–75. Popov N.V., Karnaukhov I.G., Kuznetsov A.A., et al. Epidemiological situation on plague around the world. forecast of epizootic activity of natural plague foci in the Russian Federation for 2024. Problems of Particularly Dangerous Infections. 2024;(1):67–75. DOI: https://doi.org/10.21055/0370-1069-2024-1-67-75 EDN: https://elibrary.ru/rqmbal
- Ерошенко Г.А., Куклева Л.М, Кутырев В.В. Исторические и современные классификации возбудителя чумы. Проблемы особо опасных инфекций. 2022;(4):14–22. Eroshenko G.A., Kukleva L.M., Kutyrev V.V. Historical and modern classifications of the plague agent. Problems of Particularly Dangerous Infections. 2022;(4):14–22. DOI: https://doi.org/10.21055/0370-1069-2022-4-14-22 EDN: https://elibrary.ru/jsctzk
- Ерошенко Г.А., Батиева Е.Ф., Кутырев В.В. Палеогеномика возбудителя чумы и перспективы палеогеномных исследований на территории России. Проблемы особо опасных инфекций. 2023;(2):13–28. Eroshenko G.A., Batieva E.F., Kutyrev V.V. Paleogenomics of the plague agent and prospects for paleogenomic studies in Russia. Problems of Particularly Dangerous Infections. 2023;(2):13–28. DOI: https://doi.org/10.21055/0370-1069-2023-2-13-28 EDN: https://elibrary.ru/hqaofy
- Cui Y., Yu C., Yan Y., et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl Acad. Sci. USA. 2013;110(2):577–82. DOI: https://doi.org/10.1073/pnas.1205750110
- Ерошенко Г.А., Попов Н.В, Краснов Я.М. и др. Природный мегаочаг основного подвида Yersinia pestis античного биовара филогенетической ветви 4.ANT в Горном Алтае. Проблемы особо опасных инфекций. 2018;(2):49–56. Eroshenko G.A., Popov N.V., Krasnov Ya.M., et al. Natural mega-focus of yersinia pestis main subspecies, antique biovar, phylogenetic line 4.ANT in Gorny Altai. Problems of Particularly Dangerous Infections. 2018;(2):49–56. DOI: https://doi.org/10.21055/0370-1069-2018-2-49-56 EDN: https://elibrary.ru/usvwoe
- Кутырев В.В., Попова А.Ю., Ежлова Е.Б. и др. Заболевание человека чумой в Горно-Алтайском высокогорном природном очаге в 2014 г. Сообщение 1. Эпидемиологические и эпизоотологические особенности проявлений чумы в Горно-Алтайском высокогорном (Сайлюгемском) природном очаге чумы. Проблемы особо опасных инфекций. 2014;(4):9–16. Kutyrev V.V., Popova A.Yu., Ezhlova E.B., et al. Infection of an individual with plague in the Gorno-Altaisk high-mountain natural focus in 2014. Communication 1. Epidemiological and epizootiological peculiarities of plague manifestations in the Gorno-Altaisk high-mountain (Sailyugemsky) natural plague focus. Problems of Particularly Dangerous Infections. 2014;(4):9–16. DOI: https://doi.org/10.21055/0370-1069-2014-4-9-16 EDN: https://elibrary.ru/tdyaej
- Балахонов С.В., Попова А.Ю., Мищенко А.И. и др. Случай заболевания человека чумой в Кош-Агачском районе Республики Алтай в 2015 г. Сообщение 1. Клинико-эпидемиологические и эпизоотологические аспекты. Проблемы особо опасных инфекций. 2016;(1):55–60. Balakhonov S.V., Popova A.Yu., Mishchenko A.I., et al. A case of human infection with plague in the Kosh-Agach region of the Republic of Altai in 2015. Communication 1. Clinical-epidemiological and epizootiological aspects. Problems of Particularly Dangerous Infections. 2016;(1):55–60. DOI: https://doi.org/10.21055/0370-1069-2016-4-51-55 EDN: https://elibrary.ru/vozpof
- Попов Н.В., Карнаухов И.Г., Кузнецов А.А. и др. Совершенствование эпидемиологического надзора за природными очагами чумы Российской Федерации и прогноз их эпизоотической активности на 2023 г. Проблемы особо опасных инфекций. 2023;(1):67–74. Popov N.V., Karnaukhov I.G., Kuznetsov A.A., et al. Improvement of epidemiological surveillance of natural plague foci of the Russian Federation and the forecast of their epizootic activity for 2023. Problems of Particularly Dangerous Infections. 2023;(1):67–74. DOI: https://doi.org/10.21055/0370-1069-2023-1-67-74 EDN: https://elibrary.ru/xouzbd
- Оглодин Е.Г., Ерошенко Г.А., Куклева Л.М. и др. Структурно-функциональный анализ криптических плазмид штаммов Yersinia pestis из двух природных очагов чумы России. Проблемы особо опасных инфекций. 2015;(4):82–5. Oglodin E.G., Eroshenko G.A., Kukleva L.M., et al. Tructural-functional analysis of cryptic plasmids in Yersinia pestis strains from two natural plague foci of Russia. Problems of Particularly Dangerous Infections. 2015;(4):82–5. DOI: https://doi.org/10.21055/0370-1069-2015-4-82-85
- Афанасьев М.В., Балахонов С.В., Токмакова Е.Г. и др. Анализ нуклеотидной последовательности криптической плазмиды pTP33 Yersinia pestis из Тувинского природного очага чумы. Генетика. 2016;52(9):1012–20. Afanas’ev M.V., Balakhonov S.V., Tokmakova E.G., et al. Analysis of complete sequence of cryptic plasmid pTP33 from yersinia pestis isolated in Tuva natural focus of plague. Russian Journal of Genetics. 2016;52(9):1012–20. DOI: https://doi.org/10.7868/S0016675816090022 EDN: https://elibrary.ru/wlnejp
- Ерошенко Г.А. Балыкова А.Н., Краснов Я.М. и др. Сравнительный генетический анализ штаммов Yersinia pestis, выделенных на плато Укок и других территориях Горного Алтая. Проблемы особо опасных инфекций. 2020;(4):59–69. Eroshenko G.A., Balykova A.N., Krasnov Ya.M., et al. Comparative genetic analysis of Yersinia pestis strains isolated on the Ukok plateau and other territories of the Altai Mountains. Problems of Particularly Dangerous Infections. 2020;(4):59–69. DOI: https://doi.org/10.21055/0370-1069-2020-4-59-69 EDN: https://elibrary.ru/uctrsw
- Летов Г.С. Хархира-Мунгунтайгинский участок Алтайского очага чумы. Проблемы особо опасных инфекций. 1969;6(2):37–45. Letov G.S. Kharkhira-Munguntayginsky section of the Altai plague outbreak. Problems of Particularly Dangerous Infections. 1969;6(2):37–45.
- Романова И.Ф. Шестопалов М.Ю., Балахонов С.В. Изучение дискриминирующего потенциала мультилокусного VNTR-анализа (MLVA) по выявлению межпопуляционного полиморфизма у изолятов Yersinia pestis из Тувинского и Горно-Алтайского природных очагов чумы. Журнал инфекционной патологии. 2009;16(3):186–7. Romanova I.F. Shestopalov M.Yu., Balakhonov S.V. To study the discriminating potential of multilocus VNTR analysis (MLVA) to identify interpopulation polymorphism in Yersinia pestis isolates from Tuvan and Gorno-Altaisk natural plague foci. Journal of Infectious Pathology. 2009;16(3):186–7. EDN: https://elibrary.ru/ejajpt
- Афанасьев М.В. Половинкина В.С., Балахонов С.В. и др. Использование 25-локусов VNTR-анализа для инфравидового генотипирования Yersinia pestis из Горно-Алтайского и Тувинского природных очагов чумы. В кн.: Молекулярная диагностика — 2010: сборник трудов VII Всероссийской научно-практической конференции с международным участием. Том 1. М.;2010:361–3. Afanas'ev M.V. Polovinkina V.S., Balakhonov S.V., et al. The use of 25 VNTR analysis loci for the infrapecific genotyping of Yersinia pestis from the Gorno-Altaisk and Tuvan natural plague foci. In: Molecular Diagnostics – 2010: Proceedings of the VII All-Russian Scientific and Practical Conference with International Participation. Volume 1. Moscow;2010:361–3.
- Балахонов С.В., Вержуцкий Д.Б., Холин А.В. и др. Тувинский природный очаг чумы. Иркутск;2019. Balakhonov S.V., Verzhutsky D.B., Kholin A.V., et al. Tuva Natural Plague Focus. Irkutsk;2019. EDN: https://elibrary.ru/aczoxn
- Вержуцкий Д.Б., Ткаченко С.В., Галацевич Н.Ф. и др. Обнаружение новых эпизоотических участков в Тувинском природном очаге чумы. Национальные приоритеты России. 2016;(4):17–21. Verzhutskiy D.B., Tkachenko S.V., Galatsevich N.F., et al. New epizootic areas detection in Tuvan plague natural focus. Russia's National Priorities. 2016;(4):17–21. EDN: https://elibrary.ru/raiksb
- Вержуцкий Д.Б., Базанова Л.П., Вержуцкая Ю.А. Эпизоотологическое значение массовых видов блох длиннохвостого суслика в природных очагах чумы. Байкальский зоологический журнал. 2020;28(2):105–9. Verzhutsky D.B., Bazanova L.P., Verzhutskaya Ju.A. Episootological significance of fleas — common parasites of longtailed ground squirrels in natural plague foci. Baikal Zoological Journal. 2020;28(2):105–9. EDN: https://elibrary.ru/fyaafl
- Vogler A.J., Chan F., Wagner D.M., et al. Phylogeography and molecular epidemiology of Yersinia pestis in Madagascar. PLoS Negl. Trop. Dis. 2011;9(5):e1319. DOI: https://doi.org/10.1371/journal.pntd.0001319
- Балахонов С.В., Ярыгина М.Б., Гладких А.С. и др. Молекулярно-генетическая характеристика штаммов Yersinia pestis, выделенных на монгольской территории трансграничного Сайлюгемского природного очага чумы. Проблемы особо опасных инфекций. 2019;(3):34–42. Balakhonov S.V., Yarygina M.B., Gladkikh A.S., et al. Molecular-genetic characteristics of Yersinia pestis strains isolated in the Mongolian territory of transboundary Sailyugem natural plague focus. Problems of Particularly Dangerous Infections. 2019;(3):34–42. DOI: https://doi.org/10.21055/0370-1069-2019-3-34-42 EDN: https://elibrary.ru/mlygjw
- Ярыгина М.Б., Корзун В.М., Балахонов С.В. и др. Генотипическая структура Yersinia pestis ssp. central asiatica biovar altaica в Горно-Алтайском высокогорном природном очаге чумы при MLVA25-типировании. Проблемы особо опасных инфекций. 2021;(2):138–49. Yarygina M.B., Korzun V.M., Balakhonov S.V. MLVA25-typed Yersinia pestis ssp. central asiatica biovar Altaica genotype structure in Gorno-Altai mountain natural plague focus. Problems of Particularly Dangerous Infections. 2021;(2):138–49. DOI: https://doi.org/10.21055/0370-1069-2021-2-138-147 EDN: https://elibrary.ru/hqwoiw
- Горюнова П.А., Куклева Л.М. Балыкова А.Н. и др. MLVA25- и CRISPR-генотипы штаммов Yersinia pestis из Прикаспийского песчаного очага чумы. Проблемы особо опасных инфекций. 2023;(4):68–76. Goryunova P.A., Eroshenko G.A., Balykova A.N., et al. MLVA25 and CRISPR genotypes of Yersinia pestis strains from the Caspian sandy plague focus. Problems of Particularly Dangerous Infections. 2023;(4):68–76. DOI: https://doi.org/10.21055/0370-1069-2023-4-68-76 EDN: https://elibrary.ru/mzqwsh
- Li Y., Cui Y., Hauck Y., et al. Genotyping and phylogenetic analysis of Yersinia pestis by MLVA: Insights into the worldwide expansion of Central Asia plague foci. PLoS One. 2009;4(6):e6000. https://doi.org/10.1371/journal.pone.0006000
- Vogler A.J., Keys C.E., Allender C., et al. Mutations, mutation rates, and evolution at the hypervariable VNTR loci of Yersinia pestis. Mutat. Res. 2007;616(1-2):145–58. doi: 10.1016/j.mrfmmm.2006.11.00722
- Коврижников А.В., Балыкова А.Н., Шевченко К.С. и др. Программа для ЭВМ «FramgentFinder v0.4: программа для поиска фрагментов в бактериальном геноме». Свидетельство №2024668532;2024. Kovrizhnikov A.V., Balykova A.N., Shevchenko K.S. et al. The computer program "FramgentFinder v0.4: a program for searching fragments in the bacterial genome". Certificate No. 2024668532;2024.
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. DOI: https://doi.org/10.1093/nar/27.2.573
- Selander R.K., Caugant D.A., Ochman H., et al. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl. Environ. Microbiol. 1986;51(5):873–84. DOI: https://doi.org/10.1128/aem.51.5.873-884.1986
- Hunter P.R., Gaston M.A. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J. Clin. Microbiol. 1988;26(11):2465–6. DOI: https://doi.org/10.1128/jcm.26.11.2465-2466.1988
- Никифоров К.А., Оглодин Е.Г., Макашова М.А. и др. Разработка комплексной системы молекулярно-генетической идентификации штаммов Yersinia pestis. Проблемы особо опасных инфекций. 2023;(1):126–31. Nikiforov K.A., Oglodin E.G., Makashova M.A., et al. Development of an integrated system for molecular-genetic identification of Yersinia pestis strains. Problems of Particularly Dangerous Infections. 2023;(1):126–31. DOI: https://doi.org/10.21055/0370-1069-2023-1-126-131 EDN: https://elibrary.ru/drdorj
- Li J., Wang Y., Liu F., et al. Genetic source tracking of human plague cases in Inner Mongolia-Beijing, 2019. PLoS Negl. Trop. Dis. 2021;15(8):e0009558. DOI: https://doi.org/10.1371/journal.pntd.0009558
- Zuo X., Liu F., Hu Y., et al. Genomic diversity and transmission patterns of Yersinia pestis in Inner Mongolia Autonomous Region, China. Commun. Biol. 2024;7(1):1480. DOI: https://doi.org/10.1038/s42003-024-07190-6
Supplementary files

