


Vol 101, No 5 (2024)

- Year: 2024
- Published: 18.11.2024
- Articles: 15
- URL: https://microbiol.crie.ru/jour/issue/view/186

Congratulations of the centenary of the "Journal of Microbiology, Epidemiology and Immunobiology"

 579-580
579-580



ORIGINAL RESEARCHES
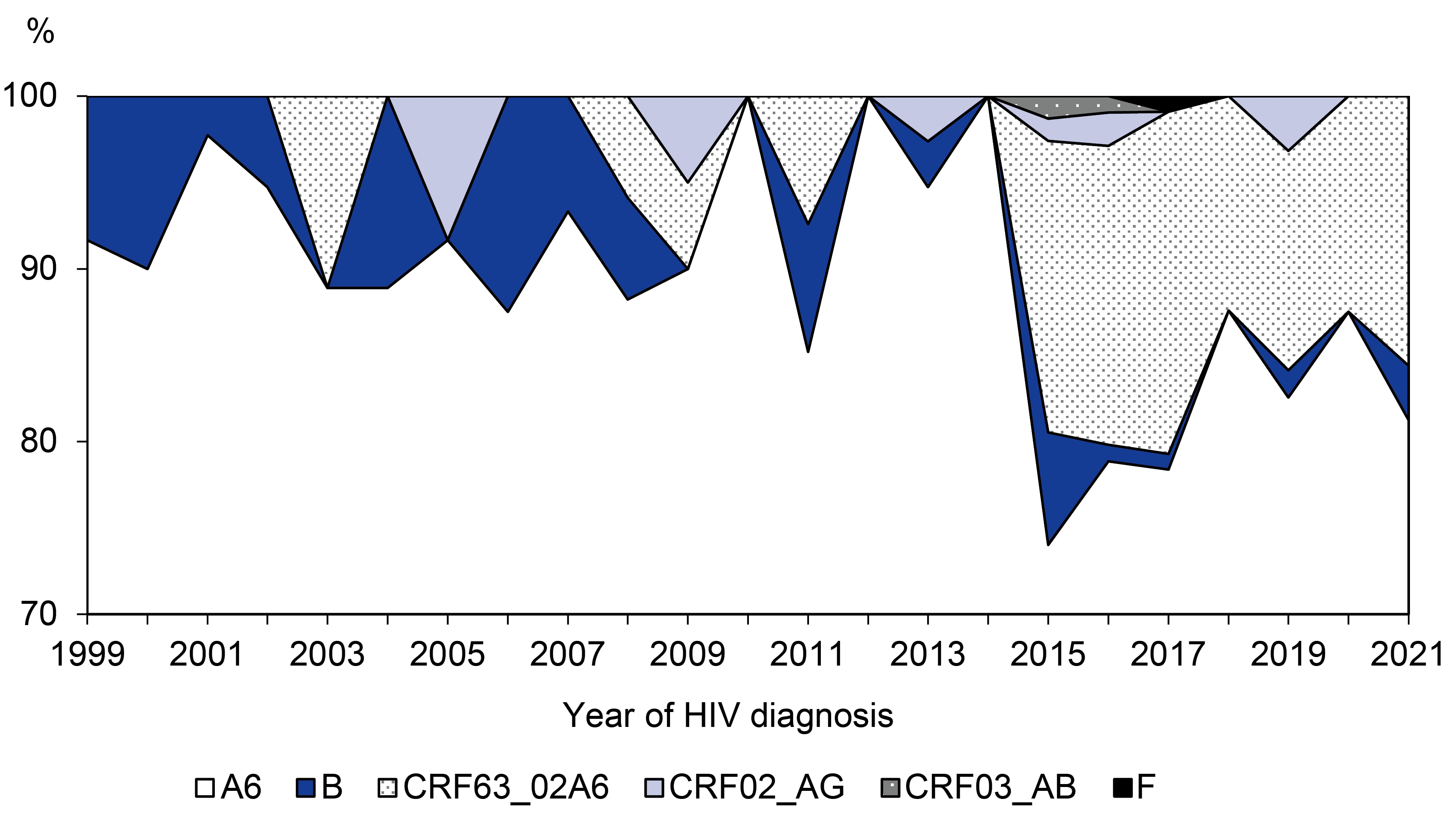
The pilot study of the features of HIV-1 resistant variants spread using molecular clusters
Abstract
Introduction. As a result of routine testing of HIV-1 drug resistance (DR), a significant amount of viral nucleotide sequences and epidemiological data of HIV-infected individuals have been collected. Combined with the increasing use of bioinformatics methods in practice, it has become possible to study the features of HIV-1 resistant variants spread using molecular clustering analysis.
The aim of the study was to validate the molecular clustering analysis in a pilot region of Russia using a significant number of nucleotide sequences to study the features of the spread of HIV-1 resistant variants.
Materials and methods. HIV-1 nucleotide sequences were obtained from 899 HIV-infected patients who were registered at the Oryol AIDS Center in 2016–2021. HIV-1 genetic variants were determined using the Stanford University database, REGA and HIV BLAST. Resistance mutations and prognostic HIV-1 DR were determined using the Stanford University database. Phylogenetic analysis was carried out using the MEGA program. HIV-1 molecular clusters were identified using Cluster Picker software.
Results. In the pilot region, sub-subtype A6 dominated (85.7%); an increase in the share of CRF63_02A6 was noted. HIV-1 resistance was found in 13.6% of patients without antiretroviral therapy (ART) experience and in 52.0% with ART experience. Molecular clusters were more often formed by HIV-1 nucleotide sequences from ART-naïve patients. HIV-1 DR variants were less likely to fall into molecular clusters. The sources of transmitted mutations were more often patients with ART experience. The most actively and efficiently transmitted mutations were K103N, V179E/T, Y181C and G190S, associated with virus resistance to efavirenz and nevirapine.
581-593
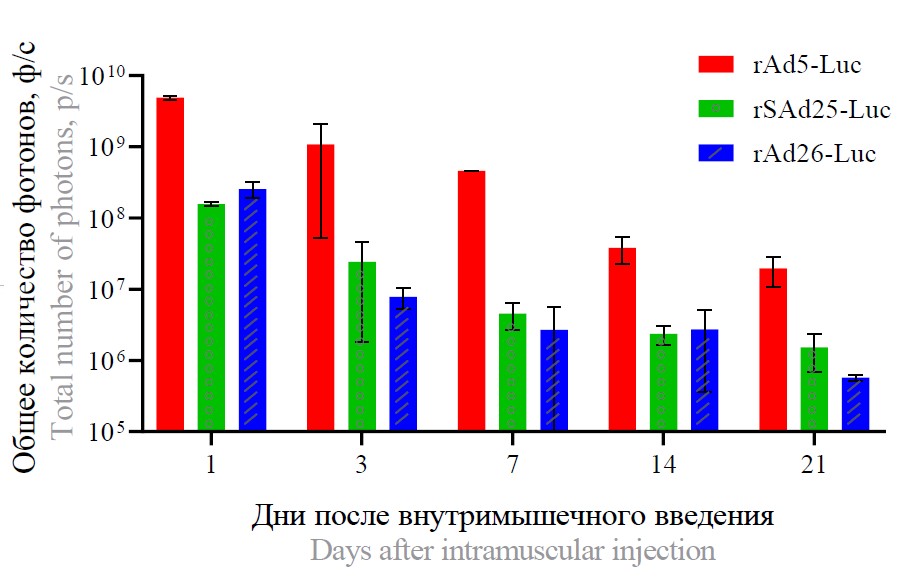
In vitro and in vivo tropism and biodistribution of recombinant simian adenovirus type 25
Abstract
Introduction. Recombinant adenoviruses are widely used in the development of vaccines for a variety of infectious diseases. Despite numerous clinical studies, only a few types of human (types 5 and 26) and simian (isolate Y25) adenoviruses are currently used to produce vaccine formulations. Different types of adenoviruses vary in their cellular tropism, which plays a key role in their ability to elicit an immune response.
The aim of this study was to investigate the cellular tropism of the simian adenovirus type 25 in vitro and its biodistribution in vivo in comparison with human adenoviruses types 5 and 26.
Materials and methods. The efficiency of in vitro transduction was evaluated on 15 different cell lines using recombinant adenovirus vectors expressing the enhanced green fluorescent protein (EGFP) reporter gene. In vivo biodistribution and bioluminescence imaging were evaluated in BALB/c mice after administration of recombinant adenoviral vectors encoding the luciferase reporter gene. The acute toxicity of a recombinant simian adenovirus type 25 vector was assessed in mice and rats following intramuscular or intravenous administration.
Results. Recombinant simian adenovirus effectively transduces a wide range of cells. At the same time, a higher tropism to human glioblastoma cells (GL-6) was found compared to the other two studied adenoviruses. In vivo experiments have shown that recombinant adenoviruses are mainly localized at the injection site, and transgene expression persists for 21 days. Acute toxicity studies demonstrated that simian adenovirus type 25 vector was well-tolerated, with no animal deaths or detectable toxic effects.
Conclusion. The new platform based on the recombinant simian adenovirus type 25 is not inferior to the existing and well-established delivery systems based on human adenovirus types 5 and 26. Due to its high level of gene transfer and favorable safety profile, the use of the simian adenovirus type 25 in medicine has the potential to offer many benefits for the development of vaccines against future infectious diseases.
594-605
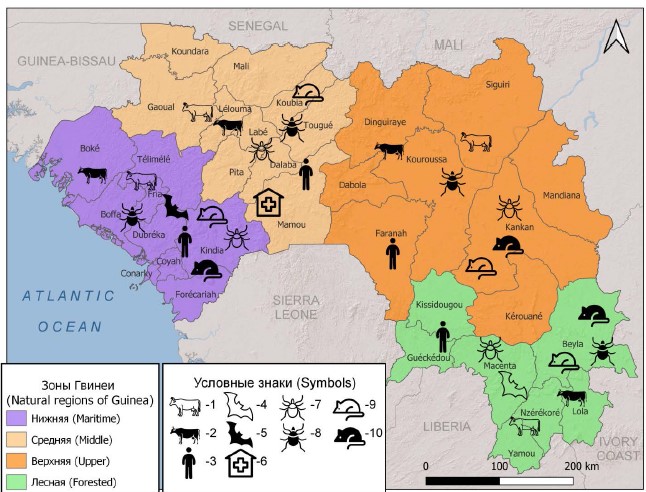
Current data on the circulation of the Q fever pathogen in the Republic of Guinea
Abstract
Background. Q fever is the one of the best-studied zoonoses, which is widespread throughout almost the entire territory of Africa, excluding the territory of the Sahara. However, the current data on the incidence of coxiellosis and the circulation of Coxiella burnetii on this continent are limited and vary according different sources.
In 1980–1990, the Soviet-Guinean Research Virology and Microbiology Laboratory conducted studies to estimate the distribution of the Q fever pathogen, assess the herd immunity in humans and identify specific antibodies in the sera of livestock. However, in subsequent years, the research was suspended.
The aim of this study is to obtain up-to-date data on the distribution of C. burnetii in all landscape and geographical zones of the Republic of Guinea.
Materials and methods. The study was carried out in the laboratory of the Russian-Guinean Center for Epidemiology and Prevention of Infectious Diseases (Kindia, Republic of Guinea). The study involved 332 sera of febrile patients and 3156 sera from practically healthy volunteers, 1074 blood samples of livestock, 1648 suspensions of ticks, 319 specimens of small mammals and 298 of bats. The study was carried out using ELISA and PCR methods, selected samples were subjected to in-depth genetic analysis.
Results and discussion. The study of the distribution of C. burnetii on the territory of all landscape-geographical zones of the Republic of Guinea was carried out. For the first time, an officially registered case of human Q fever case has been identified. The role of livestock, small mammals and bats in the circulation of the pathogen has been established. It has been shown that the main vectors in Guinea are ixodid ticks of the Amblyomma variegatum, Hyalomma truncatum and Rhipicephalus decoloratus species. Employing molecular methods, C. burnetii strains carrying the QpH1 plasmid, capable of causing diseases in humans and animals were identified. For the first time, the complete nucleotide sequence of 16S rRNA of the Q fever pathogen (OQ152497–OQ152500) identified on the territory of Guinea was determined and registered in the GenBank database.
Conclusion. Taking into account the high epidemiological significance of Q fever, the study of the specifics of C. burnetii circulation in Guinea remains an urgent task. Regular monitoring and assessment of risk factors for diseases caused by coxiella are necessary for the development of an algorithm for laboratory diagnosis and recommendations for clinicians.
606-618
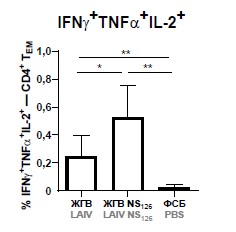
Enhancement of systemic and lung-localized CD4+ T-cell immune responses by truncation of NS1 protein of a seasonal live influenza vaccine strain
Abstract
Introduction. There is a large variety of licensed influenza vaccines worldwide, but their common limitation is rather narrow specificity and inability to protect against antigenic-drift variants of influenza virus. Therefore, optimization of immunogenic and cross-protective properties of licensed influenza vaccines is an urgent priority of public health agenda. One such approach is to modulate the immunogenic properties of live attenuated influenza vaccine (LAIV) by truncating the open reading frame of influenza virus non-structural protein 1 (NS1). The main objective of this study is to evaluate the immunogenic properties of the H1N1 seasonal LAIV strain by truncation of the NS1 protein to 126 amino acides.
Materials and methods. Using reverse genetics technique, two H1N1 LAIV strains with full-length and truncated NS1 protein with three consecutive stop codons added after the 126th amino acid residue were obtained.C57BL/6J mice were immunized intranasally with the vaccine candidates, twice at a three-week interval. Seven days after the second immunization, cells were isolated from spleen and lung tissues and stimulated with whole wild-type H1N1 influenza virus. Levels of systemic and tissue-resident cytokine-producing CD4+ and CD8+ memory T cells were assessed by intracellular cytokine staining assay with flow cytometry. Replication of engineered vaccine strains in in vitro and in vivo systems was also evaluated.
Results. Truncation of NS1 protein of the LAIV strain significantly increased the levels of virus-specific CD4+ effector memory T cells in spleens and the levels of CD4+ tissue-resident memory T cells in lungs of mice after two-dose immunization, indicating a higher potential for protection against influenza infection of the LAIV NS126 vaccine strain compared to the classical variant of LAIV. Importantly, the LAIV NS126 strain also had a more pronounced attenuated phenotype in mice than its classical counterpart.
619-627
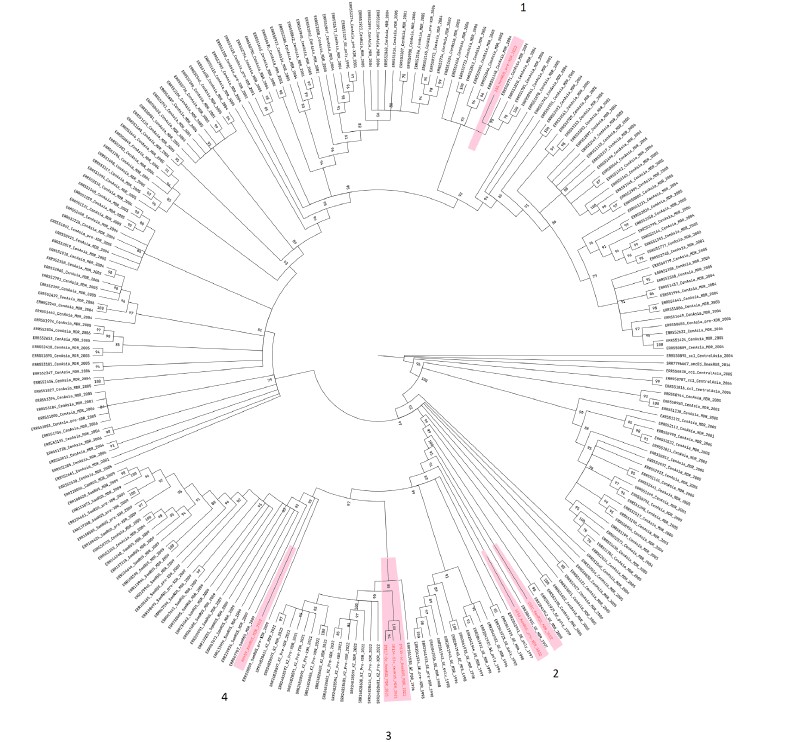
Circulation of Mycobacterium tuberculosis strains of the Beijing Central Asian Outbreak genotype in the Kemerovo region — Kuzbass in 2018–2022
Abstract
Introduction. The Kemerovo region — Kuzbass is characterized by a high prevalence of multidrug-resistant (MDR) tuberculosis (TB), including coinfection with HIV (HIV/TB). A previously unknown in Russia relationship between MDR and the Beijing Central Asian Outbreak (CAO) subtype has been discovered, which updates studies of Mycobacterium tuberculosis taking into account this resistant variant.
Objective: to study the molecular genetic structure of the M. tuberculosis population, to assess the prevalence and possible routes of emergence of Beijing CAO strains in the Kemerovo region — Kuzbass.
Materials and methods. A total of 325 M. tuberculosis strains were studied in 2018–2022 using spoligotyping, MIRU-VNTR 24 and SNP typing. Whole genome sequencing and bioinformatics analysis were performed for seven Beijing CAO strains.
Results. Primary MDR and pre-extensive drug resistance (pre-XDR) were detected in 39.4% and 11.5% of strains, respectively. In the total sample, MDR was 43.4%, pre-XDR — 19.7%. In the structure of the M. tuberculosis population, the Beijing genotype prevailed (78.8%), with its subtypes Central Asian Russian (40.9%) and B0/W148 (32.6%). The Euro-American lineage (27.3%) was represented by the genotypes T (6.5%), LAM (5.8%), Ural (4.9%), H (0.9%); one strain CAS1-Delhi was detected, the genotype of 2.8% of strains was not identified. The proportion of Beijing CAO was 12.6% of the total sample; this subtype was significantly more often detected among HIV/TB (20.6%) than in HIV-negative TB patients (9.1%; p = 0.005). The results of the Beijing CAO genome analysis from the Kemerovo region indicate the absence of a direct chain of transmission between these TB cases. A hypothesis has been put forward about the introduction of Beijing CAO to the Kemerovo region from Central Asia and its endemic circulation in the region.
Conclusion. A high level of MDR and pre-XDR was detected in Beijing genotype strains in the M. tuberculosis population of the Kemerovo region — Kuzbass, especially the B0/W148 (97.2%) and CAO (87.5%) subtypes. Beijing CAO strains, detected mainly in newly diagnosed HIV/TB patients, require further monitoring and control of their spreading.
628-640
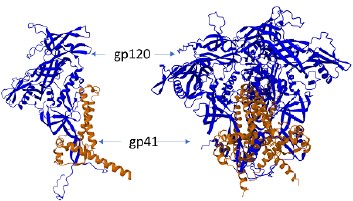
Production of a new variant of soluble trimer Env of HIV-1 CRF63_02A6 SOSIP.664
Abstract
Introduction. Obtaining stabilized recombinant HIV-1 Env trimers that have a close to the native conformation is one of the directions in the field of development of vaccines against HIV-1.
The aim of the study was to obtain and characterize the stabilized trimer Env of HIV-1 SOSIP.664 based on the circulating genetic variant of the recombinant form CRF63_02A6.
Materials and methods. Bioinformatics resources were used to design the trimer Env gene based on the HIV-1 recombinant genetic variant CRF63_02A6. The designed gene was synthesized and cloned as part of an integration plasmid vector. A stable producer of trimer Env was obtained by transfection of the CHO-K1 cell line using the developed plasmid vector. Purification of the protein complex was performed using affinity chromatography and gel filtration. Antigenic properties of trimer Env were studied using immunochemical analysis using broadly neutralizing HIV-1 monoclonal antibodies (bnAbs).
Results. A new variant of the stabilized trimer Env of the surface glycoprotein of the recombinant form CRF63_02A6 HIV-1 SOSIP.664 was designed, including additional stabilizing modifications. Based on the CHO-K1 cell line, a stable producer was obtained and a purification protocol for the designed trimer Env was developed. It was found that the trimer Env CRF63_02A6 SOSIP.664 is effectively recognized by bnAbs 2G12, VRC01 and PGT126.
Conclusion. The obtained results indicate the prospects for further study of the structural features of the trimer Env CRF63_02A6 SOSIP.664, as well as its immunogenicity and the possibility of using it as a vaccine antigen.
641-649
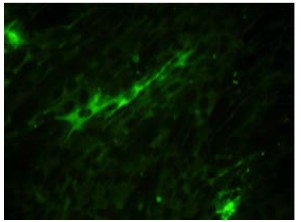
Self-replicating recombinant virus-like particles of lentivirus proliferating in glioblastoma cells and normal human macrophages
Abstract
Introduction. Both attenuated and inactivated vaccines are used in disease control. Inactivated vaccines are very diverse and include whole cell and acellular vaccines containing protein target antigens or nucleic acids encoding target antigens. Immunity induced by inactivated vaccines is not believed to be long-lasting. It is very problematic to develop a vaccine against viruses that integrate into the genome of the host cell, as well as against persistent viruses that penetrate the central nervous system (CNS), which is typical for the human immunodeficiency virus type 1 (HIV-1).
Aim of the study: to evaluate the possibility of forming HIV-1 recombinant virus-like particles (recVLPs) and HIV-1B recombinant VLPs and simian immunodeficiency virus (SIV) — SHIV89.6P based on self-replicating RNAs (srRNAs) producing target lentivirus antigens on the alphavirus replicon platform (Sindbis virus or Venezuelan equine encephalomyelitis virus (VEEV)), and also to evaluate the ability of HIV-1B and SHIV89.6P VLPs to infect glioblastoma cells and normal human macrophages.
Materials and methods. BHK-21 cells were transfected with the srRNA mixture by electroporation. Recombinant virus-like particles (recVLP’s) in recVLP’s-infected cells were detected using the immunofluorescence assay (ELISA) and electron microscopy. recVLP’s were used to infect glioblastoma cells and normal macrophages from a healthy donor.
Results. Based on the genomic RNA of the alphavirus, the plasmids were created, transcription from which makes it possible to obtain RNA that expresses lentiviral gene products in cells in quantities sufficient for the formation of mature VLPs. In BHK-21 cells infected with recVLP’s, virus-specific antigens are detected only in the cytoplasm, but not in the nucleus. Both glioblastoma cells (U87) and normal human macrophages containing CD4 receptor and SSR5 and CXR4 co-receptors give infectious progeny of HIV-1B and SHIV89.6P recVLP’s when infected with supernatant obtained after transfection of BHK-21 cells with srRNA.
Discussion. The results obtained show the possibility of expressing lentivirus structural proteins in glioblastoma cells (U87) and in normal human macrophages and can be used in the future to study the presentation of antigens in native and functional conformations in appropriate model systems to study the possibility of suppressing HIV infection in viral reservoirs in the CNS.
650-660
Production of recombinant norovirus VP1 protein and its antigenic and immunogenic properties

Abstract
Introduction. The importance of noroviruses in human infectious pathology and the danger of large epidemic outbreaks in organized groups determine the need to develop means of specific prevention of infection.
The aim of the study was to obtain recombinant norovirus VP1 protein and analyze its immunogenic and antigenic properties .
Materials and methods. Computer analysis of nucleotide and amino acid sequences, molecular cloning, polymerase chain reaction, electrophoresis of nucleic acids in agarose gel and proteins in polacrylamide gel, affinity chromatography, enzyme immunoassay.
Results and discussion. A genetic construct encoding recombinant VP1 of the GII genotype norovirus with codons optimized for highly effective expression in Escherichia coli has been created. The strain of E. coli Rosetta 2 (DE3) has been transformed by genetic construct. VP1 expression was carried out in E. coli cells, conditions for its production, purification and renaturation were optimized. A purified soluble recombinant VP1 protein forms virus-like particles with a diameter of 30–50 nm. Immunization of BALB/c mice by protein lead to antibodies production with a titer greater than 1 : 1000. When evaluating antigenic properties, it was shown that human IgG, IgM, and IgA antibodies interact with recombinant VP1. The total antibody detection rate was 47,4%. The results indicate the possibility of using recombinant VP1 for development of domestic vaccine for the prevention of norovirus infection.
661-667
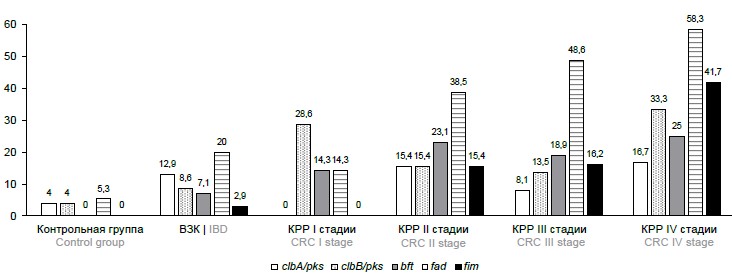
Detection of genetic determinants of potentially oncogenic representatives of the intestinal microbiota as biomarkers of colorectal cancer
Abstract
Relevance. Colorectal cancer (CRC) is the second leading cause of cancer mortality worldwide. Non-invasive diagnostic methods based on the determination of hidden blood in the stool (fecal immunochemical test, guaiac test), which have been proven to be effective in clinical studies, are used for CRC screening. However, a significant disadvantage of the available non-invasive diagnostic methods is the low sensitivity in detecting the oncological process at the early stages. A number of recent studies discuss the relationship between the disease and various potentially oncogenic microorganisms in the human intestinal tract, which can be used to expand the arsenal of non-invasive methods for diagnosing CRC based on molecular genetic examination of a stool sample to identify oncogenic microorganisms.
The aim of this study was to evaluate the possibility of using genetic determinants of potentially oncogenic microorganisms as markers for colorectal cancer, based on a comparison of their prevalence in groups of patients with colorectal cancer, facultative precancerous diseases and patients without intestinal pathology.
Materials and methods. 215 participants were included in the "case–control" study: 70 patients with newly diagnosed colorectal cancer, 70 patients with inflammatory bowel disease, 75 participants without diagnosed intestinal pathology. Polymerase chain reaction (PCR) was used to identify and detect genes of potentially oncogenic microorganisms.
Results and discussion. An association was found between CRC and the presence of the Bacteroides fragilis fragilisin gene (OR 7.00; 95% CI: 2.55–22.50; p < 0.001), species-specific genes of the periodontal pathogenic microorganisms Fusobacterium nucleatum (OR 5.61; 95% CI: 2.87–11.30; p < 0.001) and Porphyromonas gingivalis (OR 16.3; 95% CI: 4.33–106.00; p < 0.001), the clbB gene of pks pathogenicity island of the Enterobacteria (OR 3.44; 95% CI: 1.39–8.51; p = 0.010).
Conclusion. The presence of genetic markers of potentially oncogenic bacterial species and genotypes in the gut microbiome is associated with colorectal cancer. The results obtained support the possibility of using molecular genetic detection of the studied potentially oncogenic microorganisms as a method for non-invasive diagnosis of CRC.
668-678
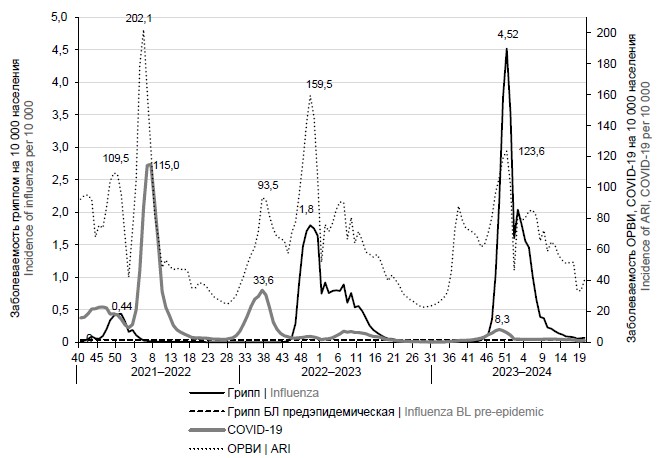
Analysis of influenza epidemics during the COVID-19 pandemic using an improved surveillance system (from 2021 to 2024)
Abstract
Aim. Assessing the effectiveness of new baselines and intensity thresholds of epidemics based on rates of incidence and hospitalization with a diagnosis of “influenza” to clarify the timing of epidemics and their spread throughout the Russian Federation against the backdrop of the COVID-19 pandemic (from 2021 to 2024).
Materials and methods. At the A.A. Smorodintsev Influenza Research Institute, the software was reformed taking into account the need to solve the tasks set during the COVID-19 pandemic. Due to changes in influenza surveillance in relation to the diagnosis of “influenza”, and hence the increase in the registration of influenza cases, the baseline and threshold of epidemics were adjusted for the influenza incidence and hospitalization rates in the surveyed cities in total (61) and for each Federal districts (over the entire population and by age groups) for 3 epidemics against the background of COVID-19 pandemic (2021–2022, 2022–2023, and 2023–2024).
Results. A comparison of baselines calculated over the previous 6 seasons based on the incidence and hospitalization rates of mostly clinically diagnosed influenza and new baseline levels of incidence and hospitalization rates of mostly laboratory-confirmed influenza during the pandemic showed minor changes in the indicators of baseline incidence and hospitalization rates while epidemic thresholds for these indicators increased.
Conclusion. Against the backdrop of the COVID-19 pandemic during the 2020–2021 season, there was no influenza epidemic. In 2021–2022, the A(H3N2) epidemic was of low intensity in terms of incidence, hospitalization rates and low mortality (2 cases). In 2022–2023, the influenza A(H1N1)pdm09 and B epidemic was of moderate intensity in terms of incidence, with a high frequency of hospitalizations and mortality (120 cases). Influenza A(H3N2) and B epidemic in 2023–2024 was of a “very high” level in terms of the influenza incidence, but average in terms of the frequency of hospitalizations and mortality (41 cases), with higher incidence rate compared to the previous epidemic (0.28% and 0.19% of the total population), including persons over 15 years of age (0.19% and 0.12%).
679-691
SCIENCE AND PRACTICE
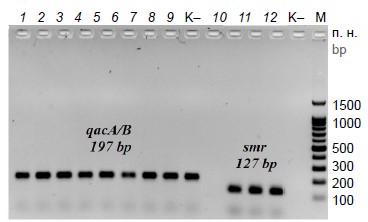
Multiplex real-time PCR for detection of qac A/B and smr genes in Gram-positive bacteria
Abstract
Background. Disinfectants are effective means of non-specific prevention of infections associated with the provision of medical care. Violation of disinfectant use regimes leads to the formation of microorganism resistance to them. To monitor the spread of clinically significant microorganisms resistant to disinfectants, the development of methods for their detection, including molecular genetic methods, remains relevant.
The aim of the study was to develop a multiplex real-time PCR for the identification of qacA/B and smr genes, the determinants of resistance to cationic biocides, in Gram-positive bacteria.
Materials and methods. Conserved regions of qacA, qacB and smr genes were searched, and primers and probes were designed using BLASTN, GeneRunner, and Multiple Primer Analyzer programs. To evaluate the analytical sensitivity of the multiplex PCR, plasmids pTZ57-qacA/B, pTZ57-smr, and pTZ57-16S containing qacA/B, smr and 16S rRNA gene fragments of 197 bp, 127 bp, and 287 bp, respectively, were constructed. The method was tested on clinical isolates of Gram-positive bacteria (n = 30).
Results. A multiplex real-time PCR using TaqMan probes was developed for the detection of qacA/B and smr genes in Gram-positive bacteria. The 16S rRNA gene was used as an internal amplification control. The sensitivity of the multiplex PCR was 103 copies for all genes. Multiplex PCR validation showed that qacA/B genes were present in 30%, smr genes were present in 10% of the isolates tested. The reproducibility of the results was 100%.
Conclusion. The developed multiplex PCR differs from existing assays by high specificity and short turnaround time, as well as by the presence of an internal amplification control. It can be used for the detection of Gram-positive bacteria potentially resistant to cationic biocides.
692-698
REVIEWS
Study of bacterial susceptibility to antibiotic and phage combinations: a literature review
Abstract
The aim of the review is to describe existing laboratory methods for determining the sensitivity of bacteria to a combination of antibiotics and bacteriophages. However, more and more often there are scientific papers in which their combined action is described as synergism. The mechanisms of this phenomenon have not been fully studied, but it has been proven that not only virulent but also moderate phages can enter into synergy with antibiotics, allowing the minimum inhibitory concentration of the antibiotic to be reduced several times. Since synergy cannot yet be empirically predicted, microbiological laboratories use various in vitro methods, most of which are labor-intensive. The development of a new technique that can be introduced into the daily practice of microbiological laboratories is relevant.
699-705
ANNIVERSARIES
90 years of the Rostov-on-Don Anti-Plague Institute: history, achievements and prospects
 706-709
706-709
CHRONICLE
Resolution of the V All-Russian scientific and practical conference with international participation "Modern immunoprophylaxis: challenges, opportunities, prospects" (Moscow, October 10–11, 2024)
710-712
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.