


Vol 99, No 6 (2022)

- Year: 2022
- Published: 15.12.2022
- Articles: 11
- URL: https://microbiol.crie.ru/jour/issue/view/53
Full Issue

ORIGINAL RESEARCHES
Molecular epidemiological study of clinical cases of acute hepatitis E in Belarus
Abstract
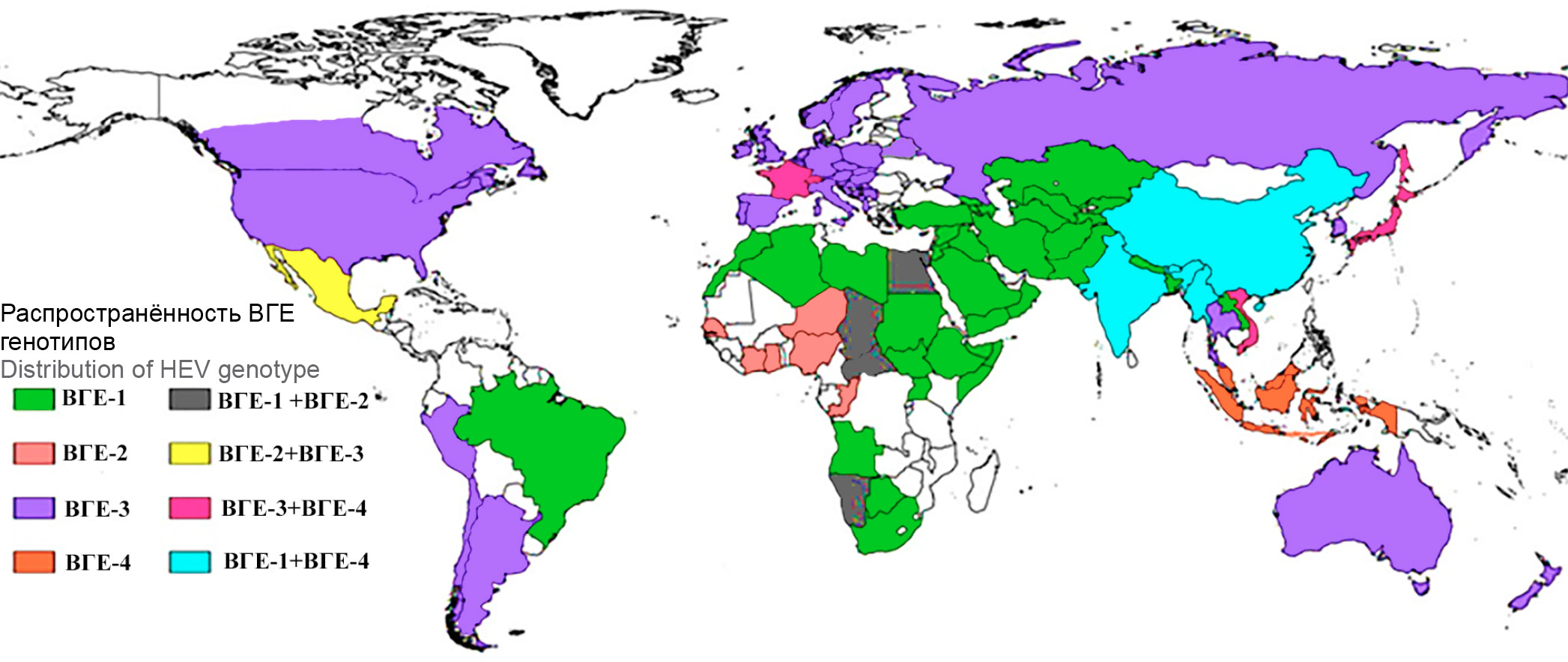
Relevance. The frequency of occurrence of anamnestic antibodies to the hepatitis E virus (HEV) in the general population of the Republic of Belarus is 7.3%, which is clearly not consistent with the low incidence of hepatitis E (HE). Most of primary HEV infections remain undiagnosed. The intensive epidemic process of HEV in the Belarusian population is hidden. Conducting epidemiological studies, including genotyping of HEV sequences isolated on the territory of the republic, makes it possible to more accurately characterize the sources of HEV infection and the mechanisms of its transmission.
Aim — molecular epidemiological study of two cases of acute hepatitis E detected in patients from Belarus.
Materials and methods. During 2021–2022, samples of biological material were obtained from two patients undergoing treatment with an established diagnosis of acute hepatitis E. Serum samples were tested to detect antibodies to HEV using enzyme immunoassay, HEV RNA was detected in fecal samples using nested RT-PCR. The nucleotide sequence was determined by an automatic sequencer using the Sanger method. Analysis of nucleotide sequences, their genotyping, and calculation of evolutionary distances were performed using MEGA X software.
Results. The HEV sequence isolated from a pregnant woman who had an epidemiological episode of alimentary contact with raw pork meat is clustered into a common phylogenetic clade with HEV sequence obtained from the patient from Belarus with a history of kidney transplantation and HEV sequences isolated from a domestic pigs. The HEV sequence isolated from a patient with a history of travel to Pakistan belongs to the HEV genotype 1 and joins a clade of HEV sequences isolated in Pakistan, India, Nepal and Mongolia.
 625-636
625-636



Molecular and genetic characterization of the hepatitis B virus full-length genome sequences identified in HBsAg-negative blood donors in Ural Federal District
Abstract
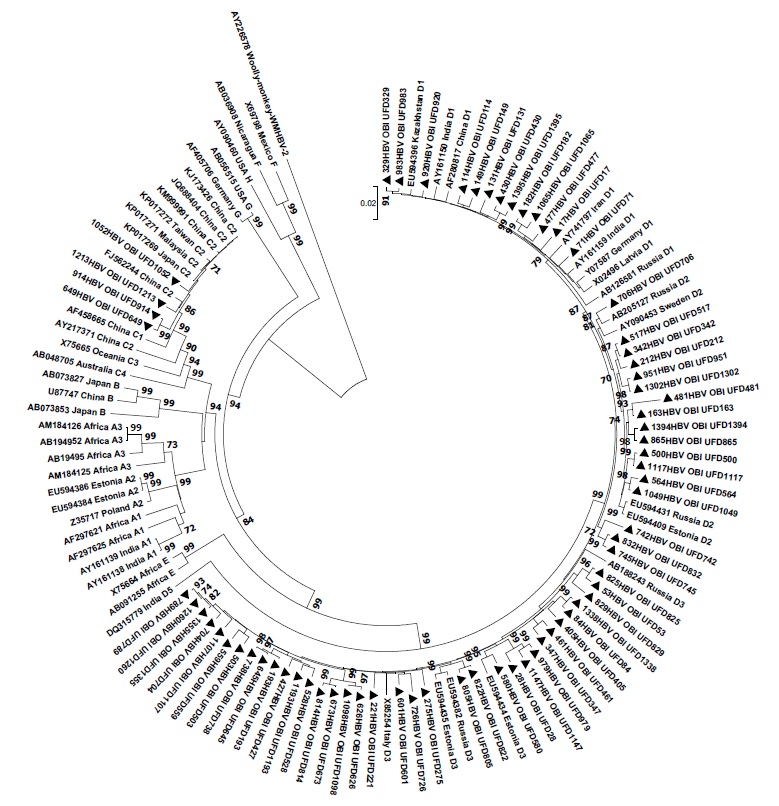
Introduction. The World Health Organization estimates that as of 2019, more than 296 million people were living with chronic hepatitis B virus (HBV) infection. The prevalence of HBsAg-negative, occult form of the disease in blood donors varies depending on the region of the world and the sensitivity of the methods of analysis used. Considering that the genetic diversity of viruses demonstrates space and time variations and taking into account that the genetic profile of isolates in key groups, which may turn into a source of the pathogen spread, is important for forecasting of the epidemiological situation, the attention should be given to identification of HBV genotypes currently circulating among regular blood donors in regions of the Russian Federation.
The aim of this work was molecular and genetic characterization of HBV genomes identified in HBsAg-negative blood donors in the Ural Federal District.
Materials and methods. The study material was 1400 plasma samples obtained from HBsAg-negative blood donors in Ural Federal District. The study included the testing for HBsAg, anti-HBs IgG and anti-HBcore IgG antibodies, HBV DNA. For all identified HBV DNA containing samples, sequencing and analysis of the nucleotide sequences of the complete HBV genomes were performed.
Results. The prevalence of HBV DNA was 4.93%, including 4 (0.28%) cases of false occult hepatitis B. Among anti-HBcore IgG-positive samples, HBV DNA was found in 18.08% of cases, while in persons with detected HBV DNA the anti-HBcore IgG positivity rate was 46.38%. In 8.69% of the isolates, anti-HBs IgG antibodies and viral DNA were detected simultaneously in the absence of anti-HBcore IgG. Based on phylogenetic analysis, HBV subgenotypes distribution in HBsAg-negative blood donors was as follows: D3 — 53.62%, D2 — 21.74%, D1 — 18.84%, C2 — 5.8%. The high variability in the S, C, P regions of the virus genome in the examined group was shown. In all cases of HBsAg-negative chronic HBV infection identified in blood donors, viral sequences contained at least one amino acid substitution in positions, mutations in which are associated with immune escape. In 3 (4.35%) cases mutations in reverse transcriptase region of P gene that are associated with resistance to the following drugs were identified: lamivudine, telbivudine, entecavir. Mutations in the preCore/Core regions that contribute to the progression of liver disease were also identified.
Conclusion. Occult HBsAg-negative chronic HBV infection poses a threat of HBV transmission through transfusion of blood and its components due to the extremely low viral load, which does not allow the virus to be detected using routinely used diagnostic kits. The situation can be exacerbated by the abundance and diversity of virus amino acid substitutions that we have identified, including immune escape mutations, drug resistance mutations, and mutations that contribute to the progression of the disease.
637-650
The effect of preventive vaccination on chickenpox incidence in Russia
Abstract
Introduction. The significance of the chickenpox (CP) problem for public health and economy of Russia necessitated inclusion of CP vaccination in the regional immunization programs of some regions of the Russian Federation and in the vaccination schedule as an epidemic-response measure.
The purpose of the study was to assess the effectiveness of CP vaccination to provide the rationale for recommendations on expansion of the national preventive vaccination schedule.
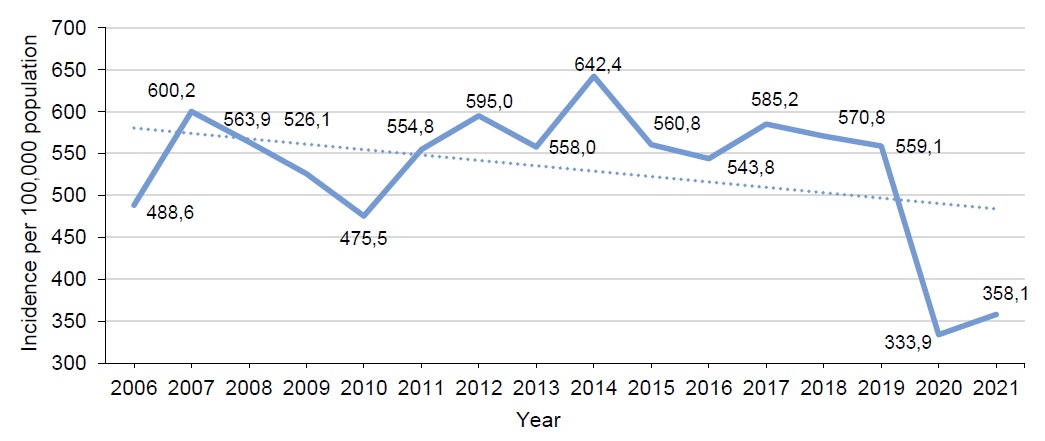
Materials and methods. The vaccination effectiveness was assessed by comparison of the vaccination rates and CP incidence rates in 2006–2021 with the reference to the data collected from forms No. 2 and No. 5 of the National Statistical Monitoring in Russia and in its regions.
Results and discussion. Before 2019, in some regions of Russia, CP vaccination of children within regional immunization programs and vaccination of risk groups within the vaccination schedule following the epidemic-response measures had hardly any effect on the epidemiological situation. The remote-work and stay-at-home policies during the COVID-19 pandemic in 2020 resulted in a decrease in the incidence and an increased number of individuals who did not have immunity against Varicella zoster, thus subsequently leading to the increased CP incidence in the country. However, the Central, Volga, and Siberian Federal Districts were able to avoid an increase in the CP incidence due to the significantly increased vaccination coverage among children in 2020–2021. At the same time, in most of the regions, less than 2% of children aged 1–6 years were vaccinated annually. The insufficient CP vaccination coverage in the regions having extensive experience of planned immunization of children led to the shift of the incidence towards older age groups and increased risk of development of congenital infection.
Conclusion. To increase the effectiveness of CP preventive vaccination, it is recommended that the national vaccination schedule should include two-dose vaccination with the coverage of at least 90% of one-year-old children, while continuing immunization of older age individuals from the groups that are at risk of infection.
651-660
Double-stranded RNAs are promising adjuvants for enhancing immunogenicity of vaccines
Abstract
Background. The most effective way to prevent infectious diseases is vaccination. Adjuvants contribute to the optimization of the immune response of vaccines. Double-stranded ribonucleic acids (dsRNAs) from natural sources are promising, but insufficiently studied adjuvants.
The aim of the work was to study the adjuvant activity of dsRNA obtained from the killer strain of Saccharomyces cerevisiae using two models of induction of a specific immune response.
Materials and methods. In the experiments, the substance of the drug Ridostin containing dsRNA, 21.72% (produced by Institute of Medical Biotechnology of the State Research Center of Virology and Biotechnology “Vector”), was used. A specific immune response was modeled using ovalbumin (OVA) or the substance of the EpiVacCorona vaccine (EVC). The experiments were carried out in 200 female BALB/c mice. Mice of the experimental groups were injected twice with antigen and adjuvant together with a 28-day interval, mice of the comparison group — with antigen only. On the 10th day after the second immunization, blood samples were collected to determine the level of specific antibodies using enzyme immunoassay. The results were evaluated by calculation of the average geometric titers of specific antibodies against OVA or EVC.
Results. OVA or EVC administered twice induced the specific antibodies in mice in dose-dependent titers. The combined administration of antigen and dsRNA increased the strength of the immune response. The highest stimulating effect of dsRNA was observed in the dose of 100 µg/mouse administered into mice immunized with OVA (1 µg/mouse) or in the dose of 50 µg/mouse in mice immunized with EVC substance (0.25 of a human dose per mouse).
Conclusion. The data obtained indicate that the substance of dsRNA exerts adjuvant properties, which gives reason to consider dsRNA as a promising adjuvant for peptide vaccines.
661-668
Competitive ability of Escherichia coli strains in the intestinal microbiota of patients with Crohn's disease and healthy volunteers: physiological, biochemical and genetic characteristics
Abstract
Introduction. Crohn's disease (CD) is a chronic inflammation of various parts of the gastrointestinal tract with an increased proportion of Escherichia coli. However, the role of E. coli in disease remains unclear.
This study aims to evaluate the competitive abilities of E. coli strains from CD patients and healthy volunteers, and to identify the biochemical and genetic determinants underlying these features.
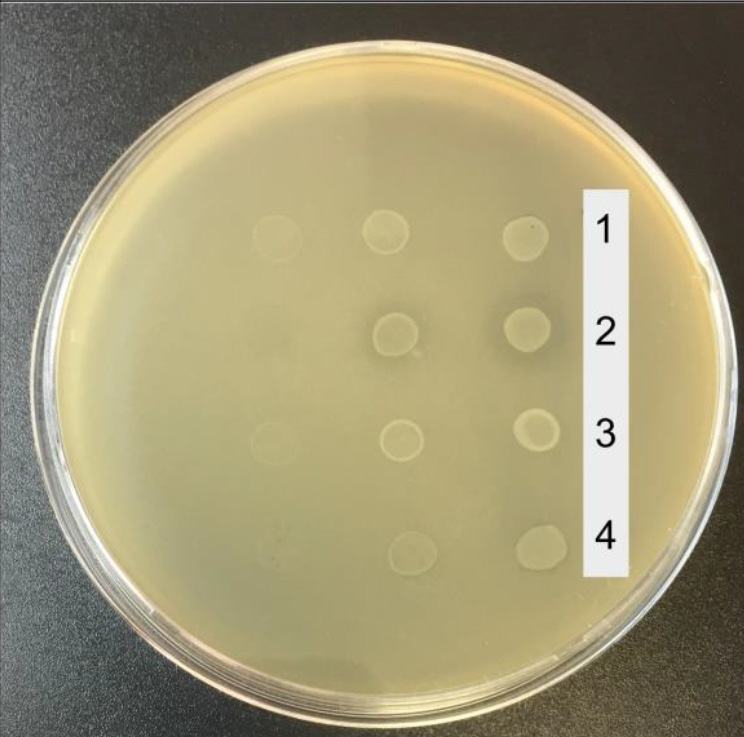
Materials and methods. The antagonistic activity was assessed by co-cultivation of 11 clinical E. coli strains inhibiting the growth of the K-12, with Enterobacter cloacae, Klebsiella pneumonia and Salmonella enterica. To elucidate the mechanism of antagonistic activity, the evaluation of biochemical properties and a comparative genomic analysis were used.
Results and discussion. Genes of bacteriocin production systems were identified in genomes of 11 strains from CD patients and healthy volunteers active against the E. coli K-12 strain. Three strains from healthy individuals demonstrated activity against several Enterobacteriaceae bacteria. The strains’ biochemical properties were typical of representatives of E. coli. Strains 1_34_12, active against E. cloacae, and 1_45_11, inhibiting all tested enterobacteria, are phylogenetically related to the laboratory strain K-12. Strain 1_39_1, active against K. pneumonia and S. enterica, is phylogenetically close to the Nissle1917, contains the genes for colibactin biosynthesis and a variant of the fimH gene that increases the adhesive ability of bacteria.
Conclusion. The identified E. coli strains are able to displace Enterobacteriaceae bacteria and can be used to study the bacteria-bacteria and host-bacteria interactions, to understand their role in gut homeostasis and intestinal inflammation.
669-681
Comparative analysis of the structure and expression of the vasH regulatory gene of type VI secretion system in toxigenic and non-toxigenic Vibrio cholerae strains

Abstract
Objective. The comparative analysis of the structure of the regulatory gene vasH of the type VI secretion system and its expression in toxigenic and non-toxigenic V. cholerae O1, biovar El Tor strains.
Materials and methods. We used 35 strains isolated from patients and from the environmental samples in the territory of Russia and Ukraine between 1970 and 2017. Analysis of the structure of the vasH gene and the amino acid sequence of the protein was carried out using Ugene 1.32, Mega X, and Bioedit v. 7.0.9.0. The relative level of vasH expression was studied by 2–ΔΔCt.
Results. The The structure of the vasH gene and the amino acid sequence of VasH protein in toxigenic typical strains and genovariants of V. cholerae O1, El Tor biovar (genotype ctxA+tcpA+) have been shown to be identical to the reference V. cholerae n16961 O1, El Tor biovar strain. The vasH sequence is variable in isolates lacking ctxA and tcpA genes (ctxA–tcpA–), and does not differ from the reference in ctxA–tcpA+ (with the exception of one strain). The studied toxigenic typical strains and the genovariants have a similar relative level of expression of the vasH gene. In isolates that do not contain the ctxA and tcpA genes, the expression of this gene is comparable to toxigenic strains, and is 3.1 times higher in ctxA–tcpA+ strains than that of ctxA–tcpA– and 2.14–2.6 times higher than that of toxigenic ones.
Conclusion. The analysis of toxigenic and non-toxigenic V. cholerae O1, biovar El Tor strains isolated in Russia and Ukraine in different periods of the current cholera pandemic confirmed the data of foreign researchers on vasH gene being intact in toxigenic isolates and variable in isolates lacking ctxA and tcpA genes. Meanwhile, the structure of vasH gene has been shown to be identical to that of toxigenic ones in 99% of the studied ctxA–tcpA+ strains. The expression of the vasH gene has been detected in all studied strains, being the highest in ctxA–TtcpA+ strains. Only two non-toxigenic strains presumably synthesizing the functionally inactive VasH protein have been identified.
682-691
Еffect of the essential oil of Satureja montana L. on the growth of cultures of conditionally pathogenic microorganisms
Abstract
Introduction. Essential oils contain antimicrobial components that are highly active against a wide range of microorganisms. Essential oils are natural, environmentally safe, low-toxic substances with a minimal list of side effects; no antimicrobial resistance is formed to them.
The aim of the research was to study the influence of the essential oil of Satureja montana L., growing in the Crimea, on the growth of cultures of opportunistic microorganisms.
Materials and methods. The short-term effect of savory oil on the growth of referenсе strains of microorganisms was studied in accordance with the European Standard for determining the rate of inactivation of microorganisms by the test substance (1997). To study the long-term effect of savory oil on clinical isolates of Staphylococcus aureus, we used the method of dilutions in a liquid medium, followed by measurement of the optical density of growth of the suspension culture biomass. The effect of savory oil on the formation of biofilms by clinical isolates of S. aureus was also studied.
Results. Whole savory oil and its dilutions of 1 : 10 and 1 : 100 with short-term action (10–60 min) completely suppressed the growth of referenсе strains of bacteria; growth of the referenсе strain Candida albicans CCM 885 was inhibited only by whole oil and a 1 : 10 dilution, while a 1 : 100 dilution had a bacteriostatic effect. Dilutions of essential oil 1 : 100 and 1 : 1000 had a pronounced antibacterial effect on the suspension culture of clinical isolates of S. aureus. Savory oil also inhibited biofilm formation by 11 isolates S. aureus.
Conclusion. The essential oil of Satureja montana L. exhibits a pronounced antimicrobial effect against referenсе strains of S. aureus ATCC 25923, Escherichia coli ATCC 25922 and fungi C. albicans CCM 885. The antibacterial effect of this essential oil on clinical isolates of S. aureus allows us to offer it as a component of combined preparations for the treatment of infections caused by antibiotic-resistant strains of staphylococcus.
701-707
Virulence determinants and genotypes of Helicobacter pylori clinical isolates
Abstract
Background. H. pylori is the principal causative agent of gastroduodenal disorders in humans. The development and severity of lesions in infected individuals depend on the virulence of H. pylori strains.
Aims: Detection of virulence determinants and comparative analysis of H. pylori genotypes in patients with chronic gastritis (CG) and duodenal ulcer (DU).
Materials and methods. The 53 H. pylori strains were isolated in St. Petersburg from patients with CG (n = 34) and DU (n = 19). The genetic determinants of virulence cagA, iceA, vacA and H. pylori genotypes in patients with CG and UC were determined using the standard PCR method.
Results. The cagA gene was found in 64.1% of H. pylori strains. The proportions of cagA+ isolates from patients with CG and DU was 55.8% (15/34) and 78.9% (15/19), respectively (p > 0.05). The iceA1 allele of H. pylori was detected in 47.4% of patients with DU, the iceA2 — in 47.1% of patients with CG (p > 0.05). The vacAs1 allele was significantly dominant in patients with DU — 94.7% versus 70.6% in CG (p < 0.05). No significant difference in vacA m1 and m2 alleles was found in H. pylori from different groups of patients (p > 0.05). All cagA+ strains were carriers of the vacA s1 allele. The vast majority of strains (10 out of 11) of the cagA–/vacAs2 genotype were isolated from patients with CG.
Conclusion. The significant association between vacAs1, vacAs2 allelic variants, as well as vacA s1/m2, vacA s2/m2 genotypes of the pathogen and severity of clinical manifestations of H. pylori infection has been established in our study. The vacAs1 and vacA s1/m2 genotypes of the pathogen are associated with duodenal ulcer.
692-700
REVIEWS
Mechanisms of Toll-like receptor tolerance induced by microbial ligands
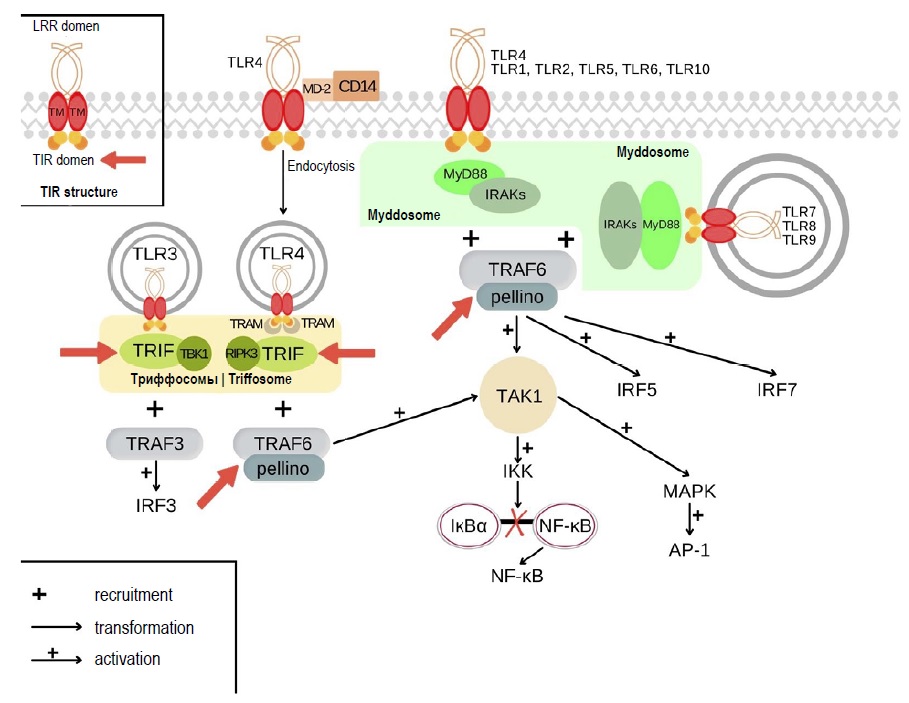
Abstract
Some microorganisms can develop tolerance. On the one hand, it allows pathogenic microbes to escape immune surveillance, on the other hand, it provides the possibility to microbiota representatives to colonize different biotopes and build a symbiotic relationship with the host. Complex regulatory interactions between innate and adaptive immune systems as well as stimulation by antigens help microbes control and maintain immunological tolerance. An important role in this process belongs to innate immune cells, which recognize microbial components through pattern-recognition receptors. Toll-like receptors (TLRs) represent the main class of these receptors. Despite the universality of the activated signaling pathways, different cellular responses are induced by interaction of TLRs with microbiota representatives and pathogenic microbes, and they vary during acute and chronic infection. The research on mechanisms underlying the development of TLR tolerance is significant, as the above receptors are involved in a wide range of infectious and noninfectious diseases; they also play an important role in development of allergic diseases, autoimmune diseases, and cancers. The knowledge of TLR tolerance mechanisms can be critically important for development of TLR ligand-based therapeutic agents for treatment and prevention of multiple diseases.
708-721
Role of intestinal microbiota in regulation of immune reactions of gut-associated lymphoid tissue under stress and following the modulation of its composition by antibiotics and probiotics administration
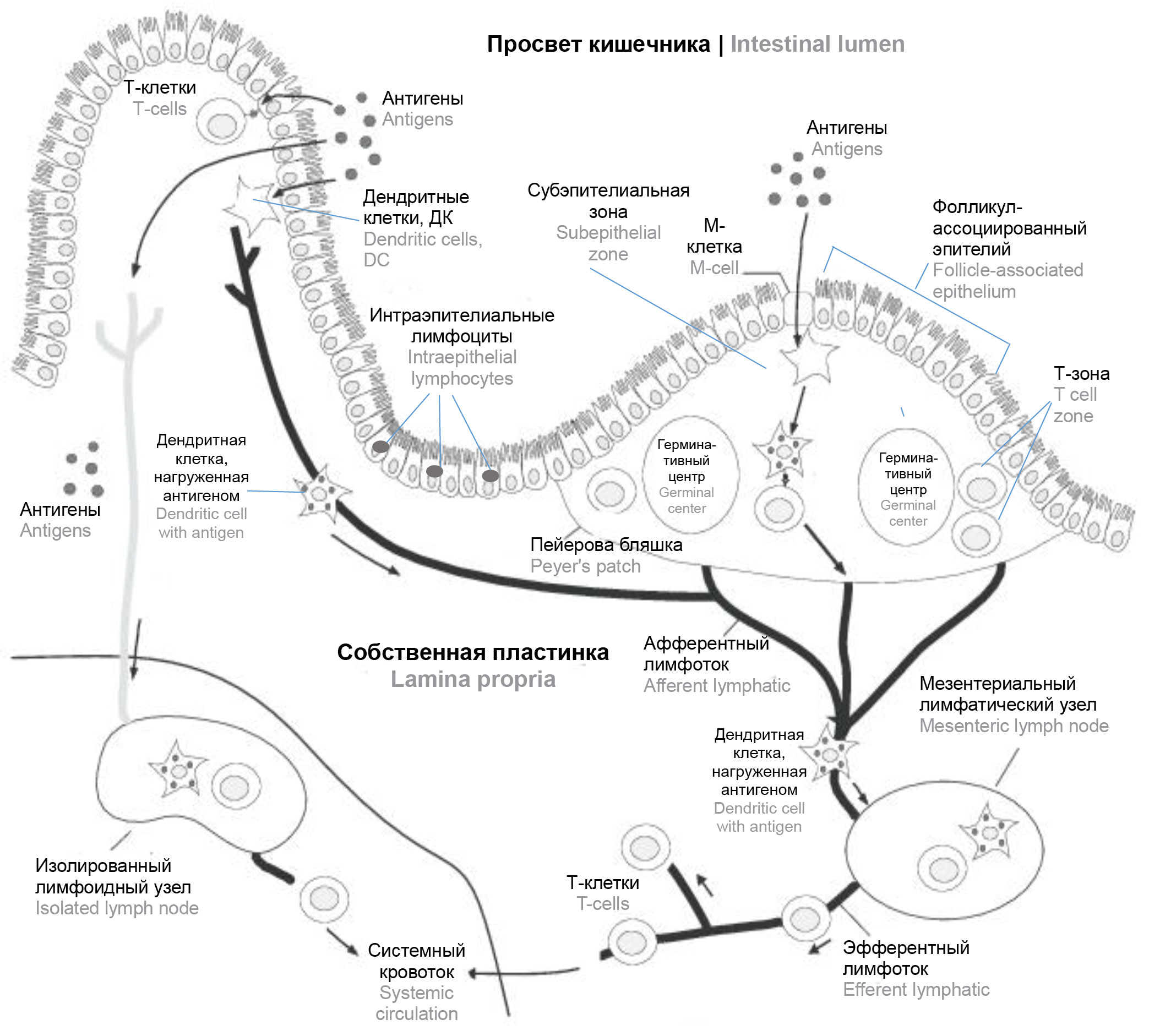
Abstract
Over the past two decades, active study of the microbial ecosystem of the host organism gastrointestinal tract has led to the recognition of gut microbiome as a "key player" that carries a significant immune pressure and is responsible both for the course of physiological processes and for the development of pathological conditions in humans and animals. A vast number of bacteria living in the human gastrointestinal tract are considered as an “organ functioning in dialogue” in formation of immunological tolerance, the regulation of normal functional activity of the immune system and maintaining the intestinal mucosa homeostasis. However, disturbances in interaction between these physiological systems is closely related to the pathogenesis of different immune-mediated diseases. In turn, in a large number of works chronic social stress, along with the use of antibiotics, pre- and probiotics, is recognized as one of the leading factors modulating in the microbiota of the gastrointestinal tract. This review focuses on the role of the gut microbiome in the regulation of immune responses of GALT under stress and modulation of its composition by antibiotics and probiotics administration.
722-733
CHRONICLE
Resolution of the XII Congress of the All-Russian Scientific and Practical Society of Epidemiologists, microbiologists and parasitologists (Moscow, October 26–28, 2022)
734-738
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.