



Journal of microbiology, epidemiology and immunobiology

The Journal is published on behalf of the Russian scientific and practical society of epidemiologists, microbiologists and parasitologists.
This Platinum Open Access peer-reviewed Journal intends to publish the cutting edge research results focused on control of spread, prevention and treatment of human infectious diseases in the field of medical microbiology, virology, epidemiology, immunology including immunodiagnostics and immunoprophylaxis.
The Journal covers the issues of human infectious diseases from the point of research of pathogen — bacteria, viruses and prions in relationship to the human immune response, intra- and interspecific interaction, environment, as well as related evolutionary processes and public health.
The Journal is intended for professionally interested physicians, epidemiologists, researchers, university lecturers, postgraduates and students.
The Journal accepts for publication Russian and foreign original research articles, reviews, short communications, mini reviews, opinion and other special featured articles as well as lectures, methodological materials related to its profile.
All papers are subject to mandatory double-blind review.
The Journal does not charge authors any fees.
JMEI belongs to the IV quartile of SJR (2022) in the specialties Epidemiology, Immunology, Immunology and Microbiology (miscellaneous), Virology and to the III quartile in the specialty Medicine (miscellaneous), is included in international bibliographic systems and international citation databases: SCOPUS, DOAJ, ULRICHS PERIODICAL DIRECT, EBSCO, OPENALEX, Fatcat, RSCI, RUSMED, ZEITSCHRIFTEN DATENBANK, GOOGLE SCHOLAR, CYBERLENINKA, RUCONT, to the recommended by Higher Attestation Commission “List of peer-reviewed scientific publications in which the main scientific results of dissertations for the degree of candidate of science should be published, competition for the scientific degree of Doctor of Science" in the following specialties:
- 03.02.02 Virology (medical and biological sciences)
- 03.02.03 Microbiology (medical and biological sciences)
- 14.02.02 Epidemiology (medical and biological sciences)
- 14.03.09 Clinical immunology, Allergology (medical and biological sciences).
In accordance with the recommendations of the Higher Attestation Commission (Letter of the Higher Attestation Commission dated December 06, 2022 No. 02-1198), the Journal belongs to the K1 category, as a publication included in the SCOPUS and RSCI databases.
The Journal follows the ICMJE's Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals.
The Journal is presented in the following online sci-hub libraries and abstract and citation databases:
- SCOPUS
- DOAJ
- ULRICHS PERIODICAL DIRECT
- Russian Science Citation Index (RSCI)
- Google Scholar
- CYBERLENINKA
- RUCONT
The most significant articles by the decision of the Editorial Board are published in full-text translation on the Journal's website under the same DOI as the original.
Each article of the Journal is assigned a digital object identifier — DOI (Crossref).
The content is available under the Creative Commons — Attribution 4.0 International, CC-BY license.
![]()
The CrossMark service is used to keep the content of the Journal up-to-date and to inform readers about changes in published articles if they occur.

The Journal uses Online First Pre-Publication — the online publication of the final version of an article that has been reviewed and accepted for publication in the Journal, without waiting for placement in a specific issue and pagination. DOI are assigned to the articles, which makes it possible to fully cite before the publication of the Journal issue.
The Editorial Board consists of 24 leading Russian and 8 foreign microbiologists, virologists, immunologists and infectious diseases doctors, including 11 full members and 6 corresponding members of the Russian Academy of Sciences.
All papers are subject to mandatory double-blind review.
The Journal is registered by the Federal service for supervision of communications, information technology and mass communications. Certificate of PI NOFS77-75442.
The Journal publishes regular issues bimonthly, six times per year.
Founders:
- Central Research Institute for Epidemiology
- Russian Scientific Society of Epidemiologists, Microbiologists and Parasitologists
Publisher:
- Central Research Institute for Epidemiology
Current Issue
Vol 102, No 5 (2025)
- Year: 2025
- Published: 05.11.2025
- Articles: 14
- URL: https://microbiol.crie.ru/jour/issue/view/192

ORIGINAL RESEARCHES
POEM: POpulation Epidemiological Model for anti-epidemic measures efficiency prognosis in the Russian Federation
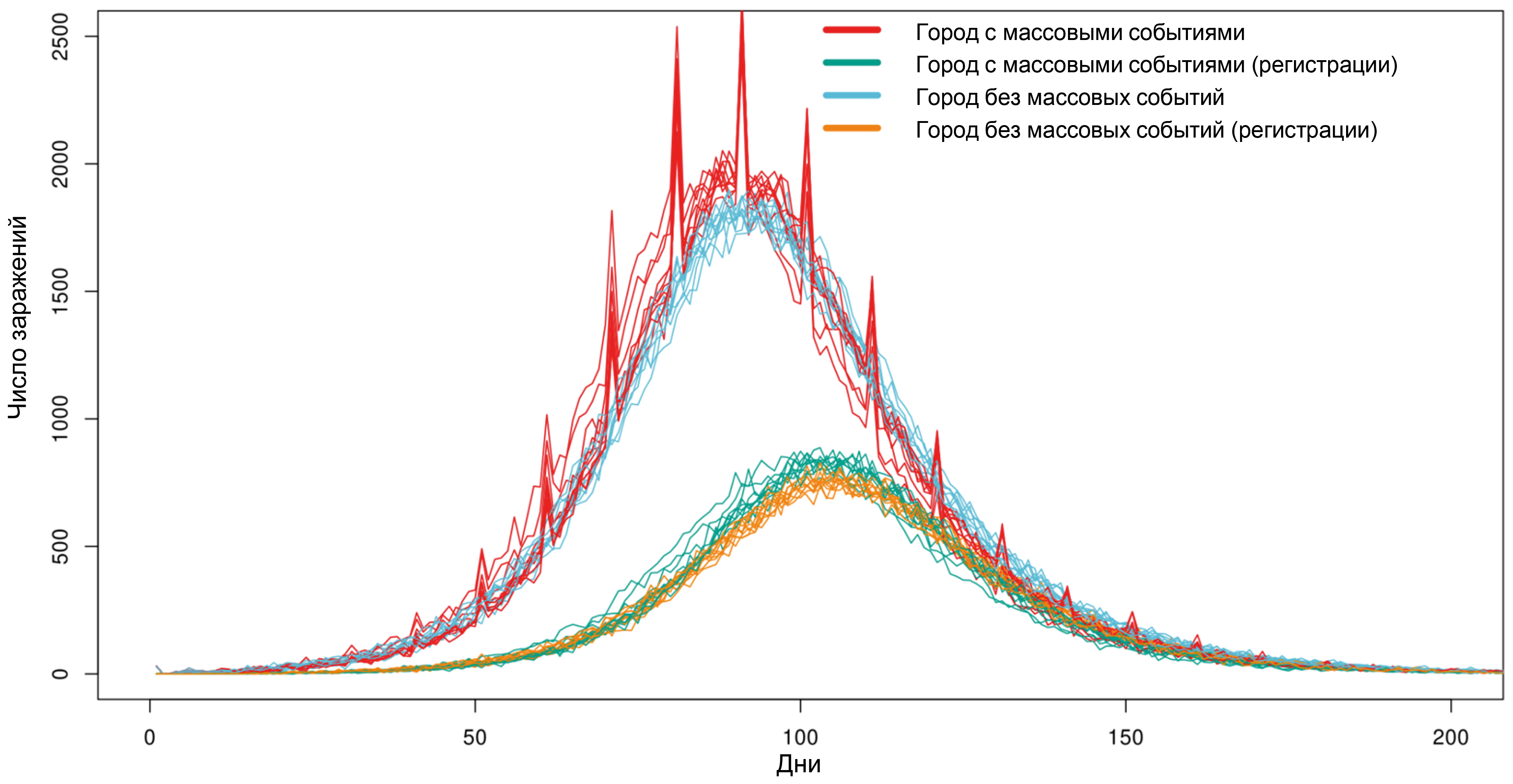
Abstract
Introduction. The COVID-19 pandemic has revealed a whole complex of problems related to mathematical modeling of the epidemic process and assessing the effect of preventive and anti-epidemic measures in modern complex societies. Along with this, the accumulation of significant factual data has spurred the active development of agent-based models, in which each agent (a hypothetical person) has a unique set of characteristics and interaction methods determined based on real sociological and demographic data.
Aim and objectives. Development and demonstration of the capabilities of the epidemiological agent-based model POEM platform (POpulation Epidemiological Model).
Materials and methods. The POEM platform is developed based on the source code of one of the most widely used agent-based models worldwide, Covasim, taking into account the demographic and organizational-administrative conditions specific to the Russian Federation.
Results. Computational experiments have shown that due to individual variability in the dynamics of infection development and the specifics of disease registration, even mass events, while leading to an actual increase in the number of infected individuals, do not have a significant impact on the shape of the curve of registered disease incidence. It has been shown that intercity traffic flows at a level of 0.1% of the population per day have a minimal effect on the dynamics of the epidemic's development, while the effect of a 1% population outflow per day sharply reduces the effect of strict anti-epidemic measures implemented in only one particular city. Using the example of the Voronezh region, the transition from the Delta variant to Omicron in early 2022 was modeled, and a high degree of correlation was shown between the model dynamics and the actual ratio of virus variants observed.
Conclusion. The model is fully implemented within the Russian system on the server of the Research Institute for System Biology and Medicine of Rospotrebnadzor and can be used to conduct digital epidemiological experiments to predict the effectiveness of proposed anti-epidemic measures.
 515-529
515-529



Genetic typing of DNA of isolates of Coxiella burnetii isolated from patients with Q fever in the Stavropol Territory
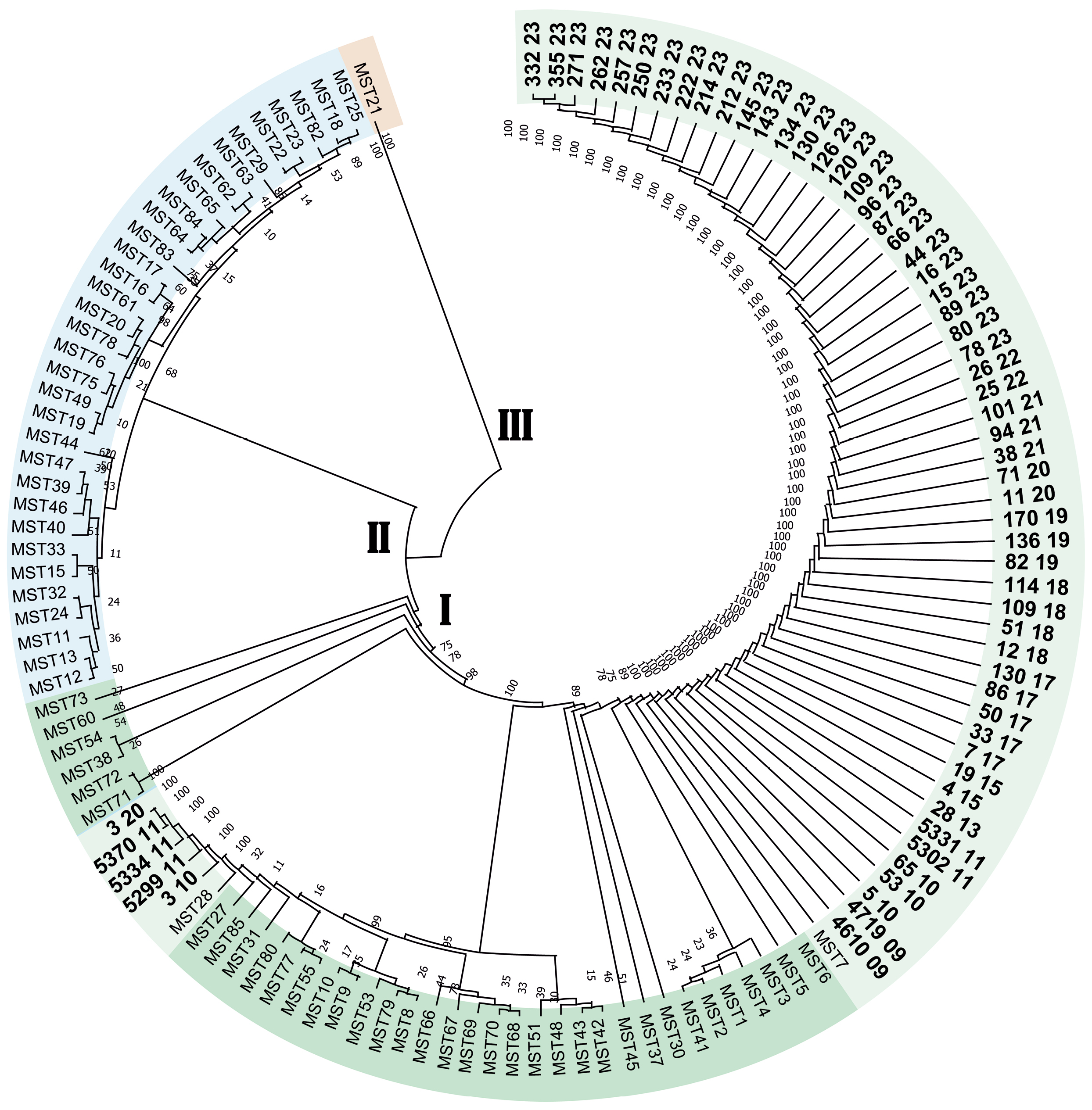
Abstract
The aim of the study is to genetically type DNA of isolates of Coxiella burnetii obtained in the Stavropol Territory (ST) from 2009 to 2023, and to analyze the genetic structure of the Q fever pathogen population in the region.
Materials and methods. The study used blood sera from febrile patients obtained from the Center for Hygiene and Epidemiology in the Stavropol Territory between 2009 and 2023. MST typing of C. burnetii was performed, and the MST type was determined using an online resource (http://ifr48.timone.univ-mrs.fr). The phylogenetic tree was constructed using the MEGA software. Plasmid typing was performed using type-specific primers for the QpH1, QpRS and QpDV plasmid loci, and the resulting amplification products were visualized by electrophoresis in a 2% agarose gel. The territorial distribution of genetic variants was analyzed using ArcGiS 10.1 software.
Results. Molecular genetic typing of C. burnetii has established that strains of the pathogen of Q fever belonging to two genotypes, MST7 and MST28 of monophyletic group I, are circulating in the Stavropol Territory. The dominant genotype in the eastern regions of the Stavropol territory is MST7, and in the northern regions, it is MST28. The pathogen of Q fever circulating in the Stavropol Territory is of a single plasmid type, QpH1.
Conclusion. Determining the plasmid and MST type allows for the genotyping of C. burnetii DNA isolates without isolating a pure culture, which can be helpful in outbreak investigations and the creation of a regional DNA isolate database.
539-546
Suppression of biofilm formation and survival of clinical isolates of uropathogens within epithelial cells under the effect of Fluorothiazinone in vitro
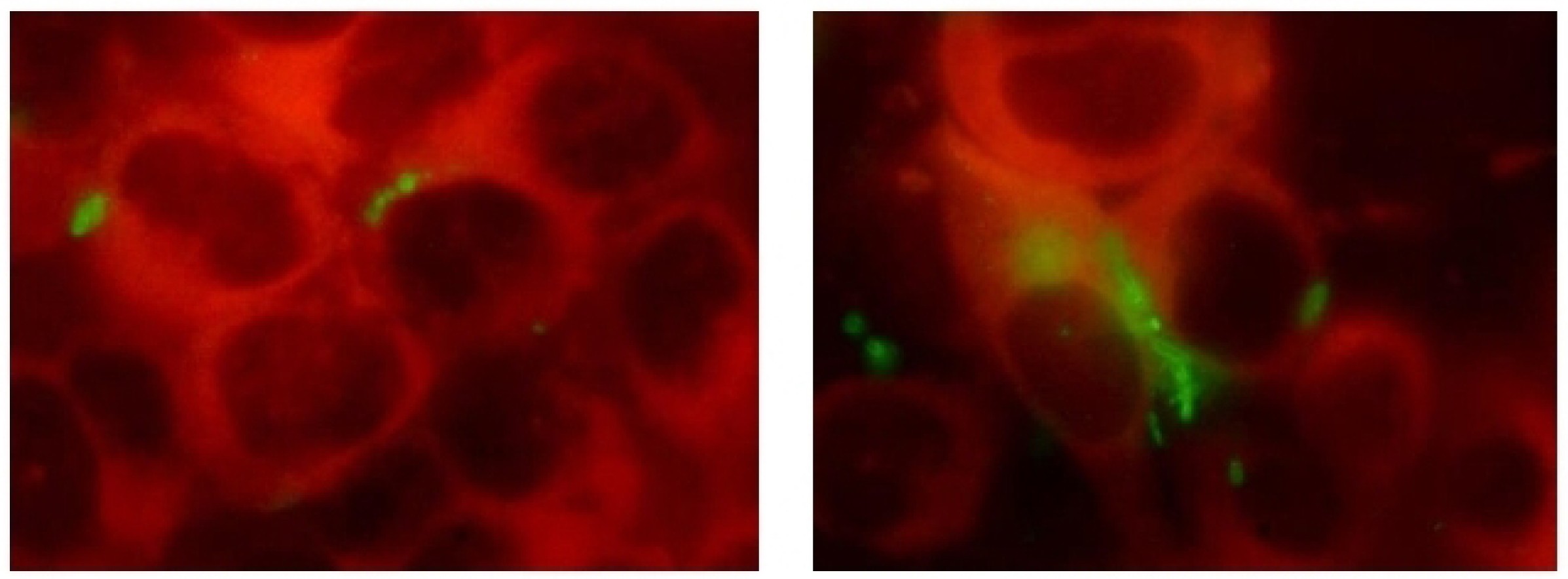
Abstract
The aim of this study was to investigate the effect of the innovative Russian antibacterial drug Fluorothiazinone, an inhibitor of the type III secretion system and flagellar function, on the virulence properties of urinary tract infection (UTI) pathogens.
Materials and methods. Clinical isolates of Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa species were obtained from 4 patients with recurrent UTI. Cytotoxicity analysis was performed on HeLa cells using the CytoTox 96 kit. Bacterial flagellar motility was assessed in 0.25% agar by measuring the zone of motility. Biofilms were cultivated in 96-well plates and stained with crystal violet and Congo red. Biofilm structure was assessed by microscopy, and the bacterial biofilm and exopolysaccharide matrix were quantified by measuring the optical density of the dye bound to the biofilm. Intracellular development was studied in PC-3 cells by determining the number of intracellular bacteria using a cell lysate plating method and an immunochemical method involving staining the bacteria with specific antibodies.
Results. The Fluorothiazinone antibacterial specifically inhibited the cytotoxicity and motility of E. coli and P. aeruginosa. For K. pneumoniae, susceptibility to Fluorothiazinone was observed, associated with the suppression of cytotoxicity. For all the isolates studied, inhibition of biofilm formation on an abiotic surface was demonstrated. For E. coli and K. pneumoniae, a significant decrease in intracellular replication within human prostate adenocarcinoma cells and suppression of intracellular bacterial community formation were observed.
Conclusion. Fluorothiazinone disrupts mechanisms that contribute to pathogen persistence in the formation of chronic UTI, including biofilm formation on abiotic surfaces such as catheters, stents, and drains, and the formation of intracellular bacterial communities and dormant intracellular reservoirs.
547-559
Association of TRIM22 gene polymorphisms (N155D and T242R) with HIV infection in the Northwestern Federal District of Russia
Abstract
Introduction. The human immunodeficiency virus (HIV) remains a global health challenge. The TRIM22 gene, which encodes a protein with antiviral activity, is a promising candidate for research, but its role in the pathogenesis of HIV infection in population of the Russian Federation has not been previously studied.
The aim of the study was to analyze the polymorphic variants rs7935564 (N155D) and rs1063303 (T242R) of the TRIM22 gene in HIV-infected individuals in the Northwestern Federal District.
Materials and methods. Polymorphic variants rs7935564 (N155D) and rs1063303 (T242R) of the TRIM22 gene were analyzed in groups of HIV-infected individuals with virological failure of antiretroviral therapy (n = 378) and practically healthy individuals (n = 319). Genotyping was performed using the polymerase chain reaction method followed by sequencing. Statistical analysis included testing for Hardy-Weinberg equilibrium of genotype distributions, assessing associations under 3 inheritance models (dominant, recessive, additive) with odds ratio (OR) and 95% confidence interval (CI) calculation, linkage disequilibrium analysis, and haplotype frequency analysis.
Results. The distribution of genotypes for the analyzed polymorphic variants was conformed to Hardy-Weinberg equilibrium expectations (p > 0.05). A significant association was found between the G allele of the rs7935564 polymorphism and the presence of HIV infection in both recessive (OR = 1.76) and additive (OR = 1.37) inheritance models. For polymorphism rs1063303, a significant association was observed only in the dominant model (OR = 1.40). Moderate linkage disequilibrium was found between the loci (D' = 0.4478; r² = 0.1572; p < 0.001). The G-G haplotype (rs7935564_G — rs1063303_G) was associated with the presence of infection (OR = 1.57).
Conclusion. In the Russian population, the polymorphic variant rs7935564 (N155D) of the TRIM22 gene is a significant genetic factor associated with HIV infection, while the results for rs1063303 (T242R) were statistically ambiguous. The association of the G-G haplotype with the presence of infection suggests a potential synergistic effect of these alleles. The data obtained highlight the importance of considering a population genetic background when evaluating the genetic determinants of the interaction between HIV and the host organism.
530-538
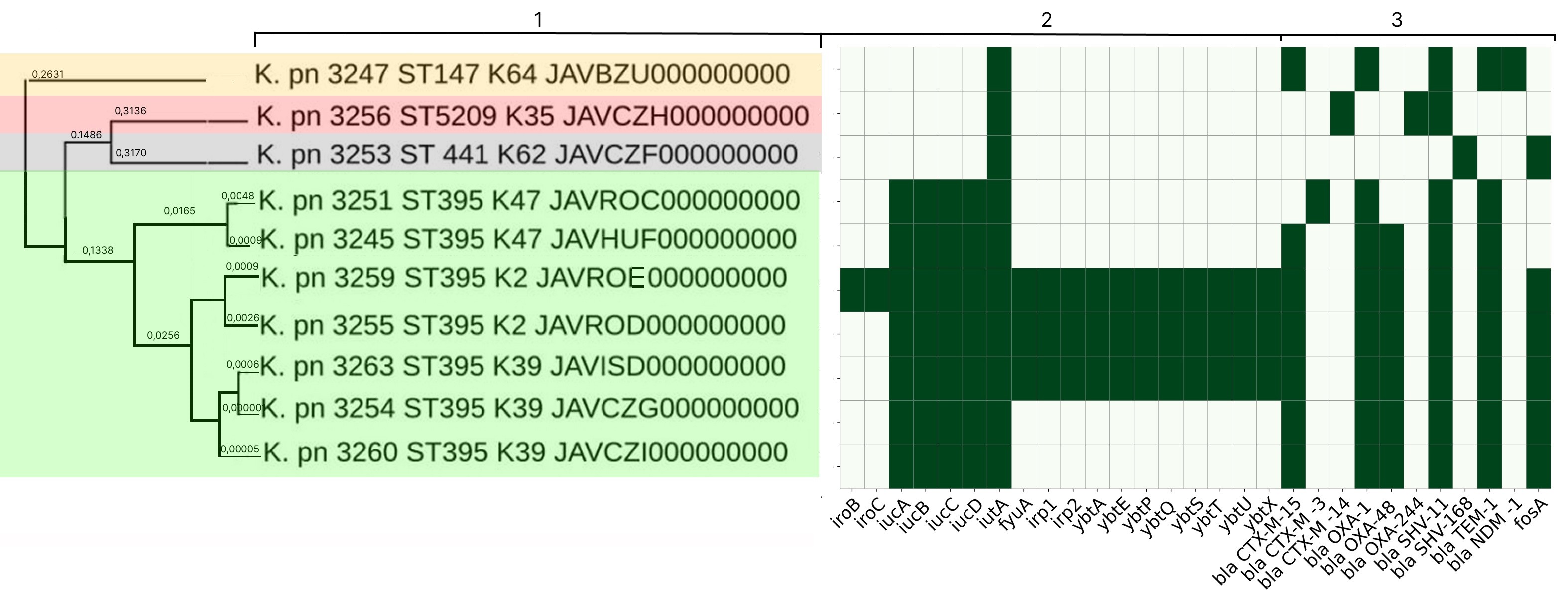
Characterization of the molecular genetic properties of epidemic strains of Klebsiella pneumoniae and Staphylococcus aureus, the pathogens of healthcare-associated infections circulating in the Nizhny Novgorod region
Abstract
Introduction. Molecular epidemiological monitoring is aimed at obtaining up-to-date information on the genetic variants of healthcare-associated infections (HAIs) circulating in the region. Currently, special attention is being paid to monitoring representatives of the ESKAPE group, as they are a frequent cause of HAIs, complicate the course of the underlying disease, and are becoming an increasingly serious threat to the health and lives of patients due to their complex pathogenicity genes and diverse antibiotic resistance mechanisms.
The aim of the study is to analyze the results of whole-genome sequencing of epidemic strains of pathogens of HAIs — Klebsiella pneumoniae ssp. pneumoniae and Staphylococcus aureus — circulating in the Nizhny Novgorod region.
Materials and methods. Classical bacteriological methods, MALDI-TOF mass spectrometry, whole-genome sequencing, and bioinformatics methods were used.
Results. In-depth analysis revealed the circulation of a population of classical K. pneumoniae strains of sequence type (ST) 3-K type (K) 3 in the neonatal intensive care unit, containing a number of virulence genes and the blaSHV-1 beta-lactamase. Circulation of a population of K. pneumoniae strains of the convergent pathotype ST395 and K39 was detected in a multidisciplinary hospital, and strains of the convergent pathotype K. pneumoniae ST395-K2, K47, as well as strains of the classical pathotype K. pneumoniae ST5209-K35, ST441-K62, ST147-K64, containing a spectrum of pathogenicity genes and beta-lactamases in their genome, including New Delhi metallo-beta-lactamase blaNDM-1, were identified. S. aureus strains associated with catheter-associated bloodstream infections have significant pathogenic potential, belonging to 13 different STs and 19 spa types (t). Circulation of methicillin-resistant (SCCmec IV, ST8, t008) and methicillin-susceptible (ST1, t127) staphylococcal strains has been detected in hemodialysis centers and departments.
Conclusion. The data obtained indicate the circulation of convergent and classical strains of K. pneumoniae and virulent strains of S. aureus in medical and preventive organizations, which justifies the need for molecular epidemiological monitoring.
560-570
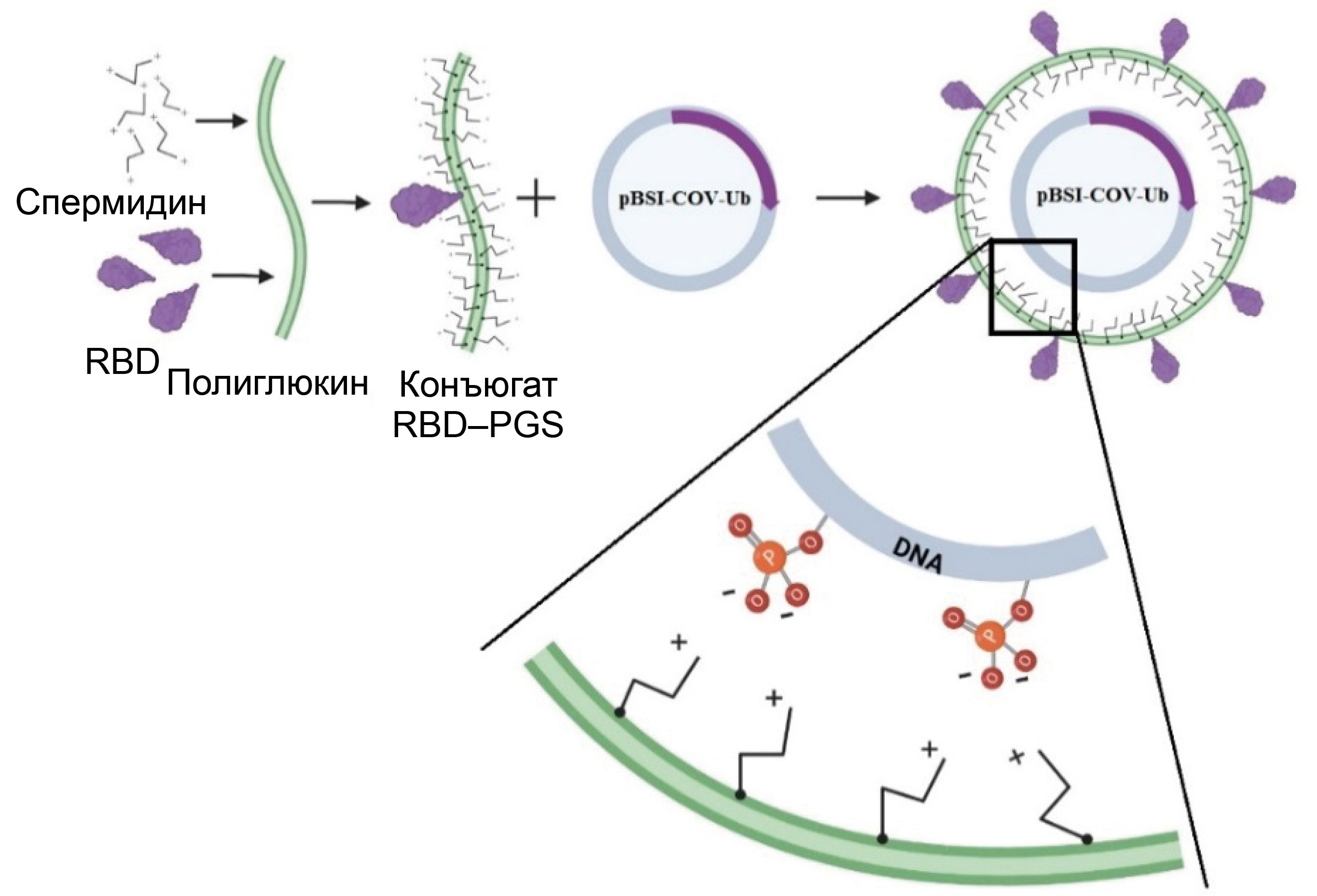
DNA-protein COVID-19 combination vaccine containing multiepitope T-cell immunogen and receptor binding domain of the SARS-CoV-2 S protein
Abstract
Introduction. During the COVID-19 pandemic, the development of preventive vaccines, including those based on new platforms, became extremely relevant. One such platform is vaccines, which combine, for example, DNA and protein components into a single vaccine.
The aim of this study was to investigate the immunogenicity of a DNA vaccine encoding a polyepitopic T-cell immunogen of the SARS-CoV-2 virus, combined with the recombinant RBD protein (the receptor-binding domain of the SARS-CoV-2 virus S protein, Wuhan-Hu-1 strain) conjugated to a polycationic carrier – polyglucin-spermidine (PGS), and to assess the contribution of individual components to the development of an immune response in BALB/c mice.
Materials and methods. To create the DNA vaccine (pBSI-COV-Ub), we used a strategy of designing an artificial polyepitope immunogen consisting of conserved immunodominant fragments of various structural proteins of the SARS-CoV-2 virus, containing a large number of T-lymphocyte epitopes: helper and cytotoxic. The recombinant RBD protein was conjugated with the polycation PGS, and upon mixing it with DNA, it formed the vaccine complex CCV–BSI, whose immunogenic properties were investigated in this work.
Results. Immunization of BALB/c mice with the CCV–BSI combined construct resulted in the induction of high antibody titers with neutralizing activity against live SARS-CoV-2 virus, as well as the formation of a virus-specific T-cell response, as demonstrated by ELISA, neutralization assay and ELISpot. It has been shown that the protein component contributes to the humoral immune response, while DNA contributes to the cellular immune response. Administration of the recombinant RBD protein led to the induction of only antibodies, administration of the DNA vaccine led to the induction of only a T-cell response, and administration of the combined preparation led to the induction of both a humoral immune response and specific T cells.
Conclusion. The unique combination of DNA and protein within a single vaccine construct allows for overcoming the limitations of each of these vaccine types and leads to the induction of both arms of immunity. The protein component can be replaced according to the current viral strain, and a universal T-cell immunogen can provide a response to a wide range of circulating variants. This platform can be further used to develop vaccines against various highly variable viruses.
571-582
Criteria for assessment of the quality of Pseudomonas aeruginosa genome sequences
Abstract
Introduction. With the development of sequencing technologies, the volume of genomic data is increasing, which necessitates the development of metrics for assessing the quality of genome assembly. Despite the unified nature of modern instruments (Plantagora, SQUAT, QUAST, BUSCO, CheckM2, etc.), they do not take into account the specific genome organization of particular species. The issue of import substitution of bioinformatics tools is particularly acute given limited access to foreign technologies. Furthermore, there are no specialized methods for assessing the quality of Pseudomonas aeruginosa genome assemblies, which is limited to general metrics (N50, number of contigs).
The aim of the study is to develop an algorithm and criteria based on a comprehensive approach for the specific assessment of the quality of whole-genome sequencing of P. aeruginosa.
Materials and methods. The study was conducted on 108 strains of P. aeruginosa. The proprietary software is developed in Java and Python languages.
Results. An algorithm for assessing the quality of P. aeruginosa whole-genome data has been developed based on the analysis of key housekeeping genes (fur, algU, dinB, etc.), genome size, GC content, and the N50 value. Genomes lacking key genes or with structural errors are classified as poor or medium, with the latter not recommended for phylogenetic analysis. The algorithm offers simple and clear parameters for assessing the quality of whole-genome data.
Conclusion. Based on the analysis of essential genes, genome size, GC content, and the N50 index, we have developed a classification of the quality of P. aeruginosa genome assemblies (good, medium, low). An algorithm and the Genomes Validator program have been created for rapid assessment.
583-591
Genetic diversity of mutations affecting the hemolytic activity of Bordetella pertussis bacteria during in vitro cultivation
Abstract
Introduction. The two-component BvgAS system regulates the transcription of the pathogen's virulence genes and plays a key role in the pathogenesis of whooping cough, an anthroponotic infectious disease. Currently, the factors affecting the BvgAS system of Bordetella pertussis bacteria in the human organism have not been studied in practice. It is known that disruption of the bvgAS operon structure leads to phase modulations and changes in virulence. IS-elements intergation into the bvgAS operon and other virulence genes of B. pertussis bacteria is important mechanism in regulating their expression, potentially leading to the long-term persistence of this pathogen in the human body.
Aim. Identification and description of spontaneous IS-elements insertions in B. pertussis into bvgAS, cya and fhaB virulence genes responsible for hemolytic activity during in vitro bacterial cultivation.
Materials and methods. B. pertussis strains from the collection of the N. F. Gamaleya National Research Center for Epidemiology and Microbiology were used: virulent B. pertussis 475 strain and its isogenic attenuated strain B. pertussis 4MKS; virulent B. pertussis strain Tohama I and its avirulent mutant B. pertussis 347. Bacteria were cultivated on casein-charcoal agar with blood addition. The formation of haemolysis zones was assessed visually. PCR, real-time PCR and sequencing were used for the genetic characterization of the obtained insertion mutants.
Results. B. pertussis mutants containing insertions of IS-elements in fhaB and bvgAS genes with impaired hemolytic activity (Hly– phenotype) have been isolated in vitro, as well as B. pertussis mutants that retained hemolytic activity (Hly+ phenotype), containing IS-elements in a previously undescribed orientation in the bvgAS gene. The frequency of insertional mutant formation depended on conditions of cultivation and bacteria genotype.
Conclusion. Arguments are made for the hypotheses about the IS-elements involvement in B. pertussis bacteria transition to a state of reduced virulence, which provides the possibility of long-term persistence of this pathogen in the human body.
592-604
Monitoring for Clostridioides difficile-associated infection in hospital
Abstract
Introduction. Clostridioides difficile — an anaerobic, spore-forming, Gram-positive bacteria — is a component of the normal intestinal microflora. C. difficile-associated infection develops during its overcolonisation, when vegetative forms produce exotoxins that cause inflammation n the colon wall. Toxigenic strains of C. difficile are the main cause of healthcare-associated infections in hospitals.
The aim of the study is to investigate the frequency of detecting C. difficile (both toxigenic and non-toxigenic strains) in patients admitted to the gastroenterology department of the Yu.M. Lopukhin Federal Scientific Clinical Center for Physical and Chemical Medicine of the Federal Medical Biological Agency of Russia in 2021–2023 with diarrhea syndrome and other established diagnoses.
Materials and methods. The study included 547 patients aged 19–88 years (46.6% male, 53.4% female). Real-time polymerase chain reaction was used to detect C. difficile DNA and its toxin A and B genes, and a bacteriological examination of stool was also performed. Upon detection of clinical signs of bacterial infection, a C-reactive protein (CRP) test was performed.
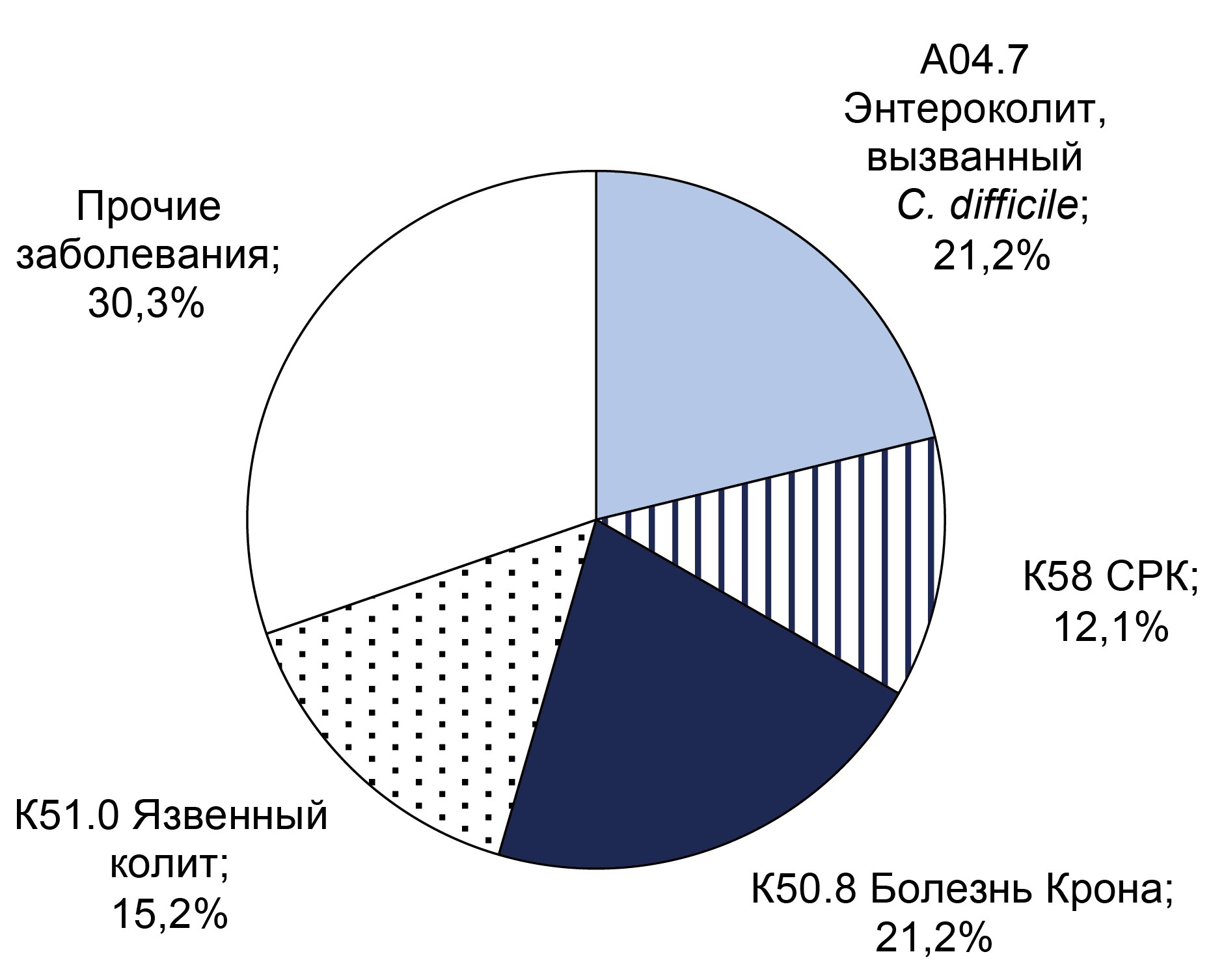
Results. C. difficile DNA was detected in 65 (11.9%) patients, and toxins A and B genes were found in 32 (5.9%) patients. Non-toxigenic strains were more frequently detected in men (55%) under 40 years, while toxigenic strains were equally frequent in both sexes under 40. CRP analysis indicated that inflammatory processes were more likely in patients over 40 years old. The predominant diagnosis in toxigenic strain carriers with high CRP was C. difficile-associated enterocolitis (ICD-10: A04.7), whereas in the non-toxigenic group, it was ulcerative colitis (ICD-10: K51). Extended bacteriological analysis revealed significant gut microbiota imbalances in all patients.
Conclusion. Over the three-year surveillance period, the prevalence of C. difficile-positive patients increased from 6.6 to 7.9%, and the proportion of samples positive for toxin A/B genes rose from 5.2% to 7.9%. These findings underscore the necessity for enhanced preventive measures to mitigate risk factors for CDI in hospital settings. Therefore, preventive measures are necessary to reduce the impact of risk factors for the development of C. difficile-associated infection in hospitals.
605-614
Improvement of the MLVA typing scheme for Burkholderia mallei strains
Abstract
Introduction. The registration of sporadic cases of glanders in horses in Russia, caused by Burkholderia mallei, highlights the importance of developing genotyping algorithms for this pathogen. The MLVA method (multilocus-variable tandem repeat analysis), based on a comparative analysis of the number of variable tandem repeats (VNTRs), remains a promising genotyping tool. As the number of whole-genome sequences in international databases increases, the informative value of VNTR loci changes, necessitating a revisiting of existing typing schemes.
The aim of this study was to assess the feasibility of including the VNTR locus BPSS1974#I in the MLVA-6 scheme for intraspecies differentiation of B. mallei.
Materials and methods. The study of 64 strains of B. mallei was conducted in silico and in vitro using MLVA, differentiating region amplification, whole-genome sequencing, and bioinformatic analysis.
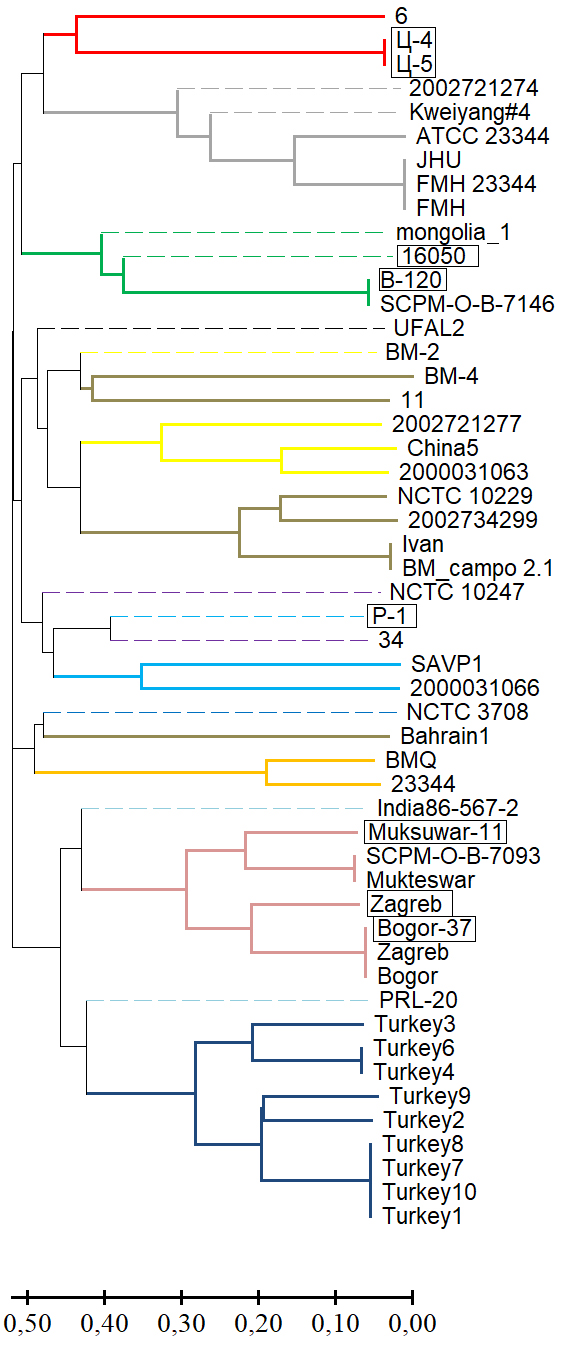
Results. Genotyping B. mallei using the MLVA-6 scheme failed to determine in silico VNTR profiles of 13 strains from the GenBank NCBI database for one or more loci due to low read coverage of the corresponding genomic regions or their complete absence (null alleles). The effective number of alleles (ne) and the polymorphic information content (PIC) index for the MLVA-6 scheme loci ranged from 3.842–8.103 and 0.740–0.877, respectively. The potential for including the VNTR locus BPSS1974#I in this scheme was determined based on the molecular stability of the motif within it and a high values for ne and PIC, which were 4.299 and 0.767, respectively. VNTR profiles of the collection strains at locus BPSS1974#I were identical to the corresponding strains in the GenBank database. The results of the cluster analysis using a combined MLVA-6 scheme and the BPSS1974#I locus were consistent with the phylogenetic reconstructions obtained using other molecular genetic methods.
Conclusion. The VNTR locus BPSS1974#I can be considered a marker, the inclusion of which in the MLVA-6 scheme will improve the accuracy of genotyping and the determination of the regions of origin of newly isolated B. mallei strains.
615-625
REVIEWS
Immunological and epidemiological effectiveness of pediatric vaccination against hepatitis A using a single dose of inactivated vaccine
Abstract
Hepatitis A is an acute liver disease caused by the hepatitis A virus (HAV), which can be prevented by means of vaccination. The standard hepatitis A vaccination schedule consists of two doses of inactivated vaccine, but for economic reasons and the purpose of improving vaccination coverage, universal pediatric single-dose vaccination programs have been implemented in certain regions of the world.
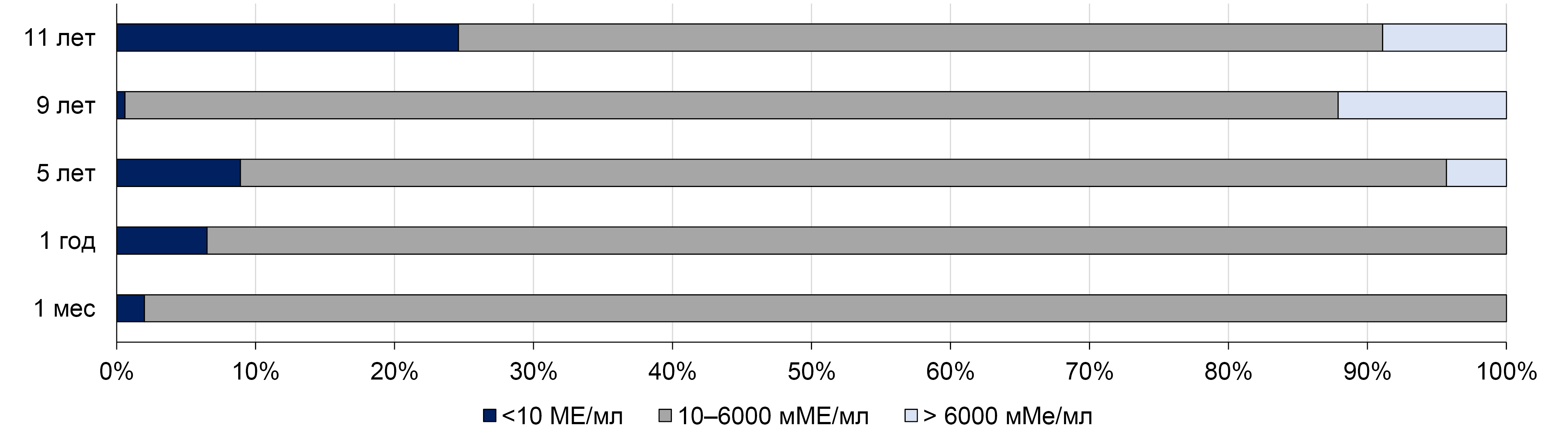
The aim of this review is to summarize and analyze published data on the duration of protective antibody levels after a single-dose pediatric immunization, as well as hepatitis A incidence in countries where hepatitis A vaccination is carried out using a single dose of inactivated vaccine.
Recent data from different regions of the world, including the Russian Federation, support the effectiveness of the single-dose hepatitis A vaccination strategy, both in terms of the duration of the immune response and the impact on the incidence rates. However, further studies on the long-term effectiveness of single-dose immunization with inactivated vaccine, as well as continuous hepatitis A surveillance, are necessary to assess the duration of protection and the necessity for booster vaccination later in life
626-634
Next-generation protective gloves: current trends in technological solutions and application prospects (scientific review)
Abstract
Introduction. Protective gloves are widely used in medicine to provide biological protection for patients and medical staff. However, if gloves are used improperly, there is a risk of healthcare-associated infections (HAIs) for both staff and patients. A significant problem is the resulting growth in medical waste and its disposal. Therefore, developing new approaches to ensure maximum protection for staff and patients, and minimize the risk of infection, by creating protective gloves based on biodegradable polymer materials with an antimicrobial coating, is an urgent epidemiological and environmental objective.
This paper discusses modern technologies and emerging issues in the creation of new materials for protective gloves with antimicrobial properties. These materials can be made using guanidine derivatives, quaternary ammonium compounds (QACs), chlorinated phenols, essential oils, iodine compounds, silver salts, metal oxides and metal nanoparticles and oxides, vegetable oil extracts, aniline dyes. The introduction of biofillers such as starch and nanocellulose will help improve biodegradability. They will also help maintain the necessary physical and chemical characteristics. The problem of synthetic rubber waste disposal can be solved by the development of new composite materials with improved biodegradation characteristics. These materials are in the form of thermoplastic elastomers (TPE), polylactide (PLA) and polylactone (PLC).
Conclusion. A review of the scientific literature revealed a significant global interest in the creation of protective gloves with antimicrobial properties made from biodegradable materials. However, in addition to directly suppressing the growth of pathogenic microflora, their use may also pose a number of problems related to their impact on human health and the ecosystem. The successful implementation of this direction hinges on the continuation of scientific research on imparting the declared properties to gloves. This research should use effective, reliable and safe technologies, as well as the development of unified methods and protocols for assessing antimicrobial activity. Once these are in place, the research can be implemented widely in practice. The production of biodegradable protective gloves offers significant potential, as it will help to reduce the risk of infection spreading in healthcare organizations and contribute to environmental protection.
635-646
ANNIVERSARIES
On the 70th anniversary of Academician of the Russian Academy of Sciences Alexander Nikolaevich Kulichenko
 647-648
647-648
CHRONICLE
Resolution of the Congress with international participation "Epidemiology — 2025" (Moscow, October 15–16, 2025)
 649-651
649-651
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.