


Vol 100, No 6 (2023)

- Year: 2023
- Published: 15.12.2023
- Articles: 11
- URL: https://microbiol.crie.ru/jour/issue/view/181

ORIGINAL RESEARCHES
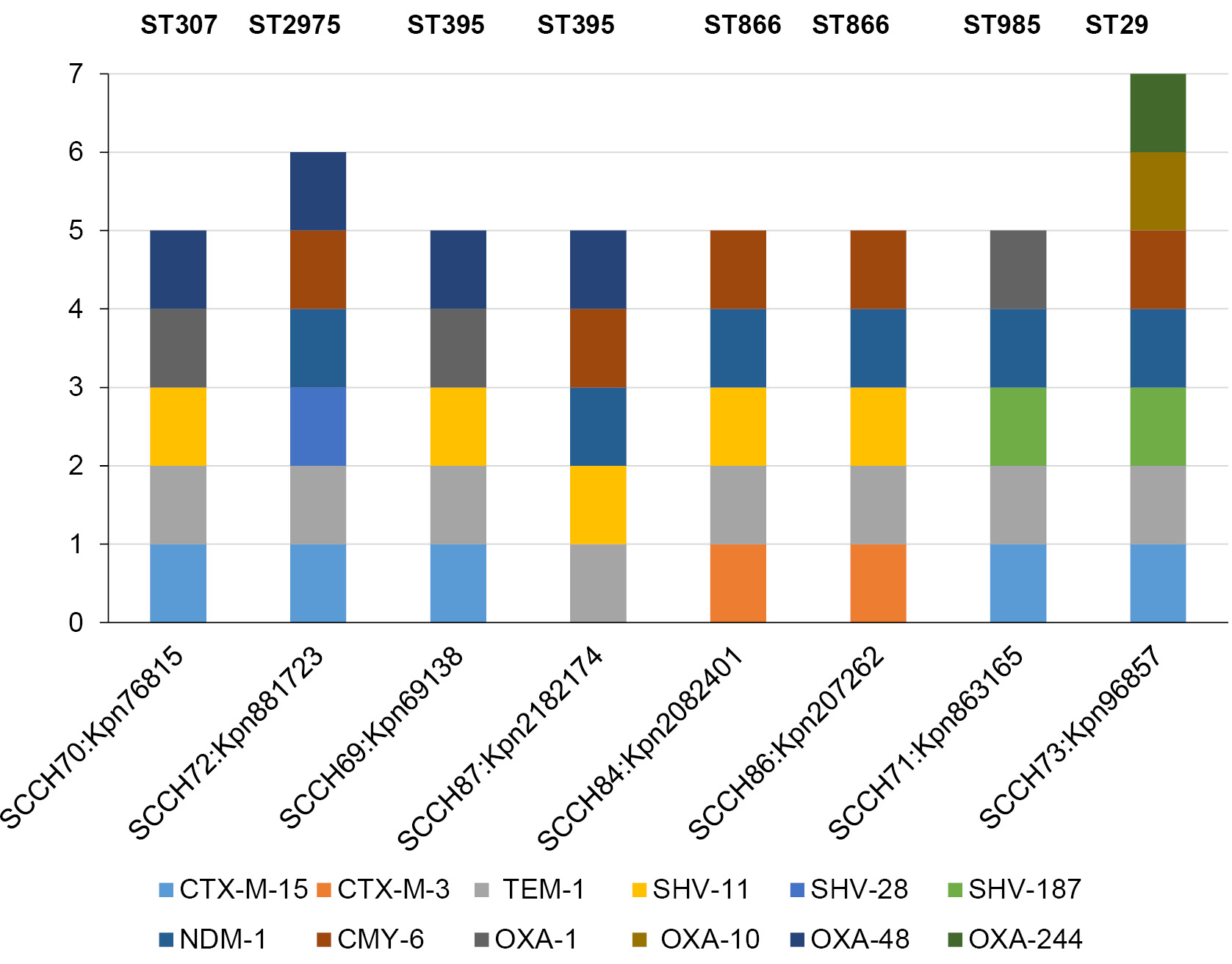
Genomic features of resistant Klebsiella pneumonia, isolated from the bloodstream and cerebrospinal fluid of pediatric hospital patients
Abstract
Introduction. Carbapenemase-producing Klebsiella pneumoniae (CP-Kp), which are international high-risk clones, have become a problem of utmost importance. CP-Kps, adapting to the hospital environment, evolve into convergent pathotypes. Such variants combine traits of two genetic lineages: multidrug resistant (MDR) and hypervirulent. The pathotypes, along with MDR K. pneumoniae, pose an exceptional threat to young patients during systemic infection.
The objective of this study is the detailed molecular genetic analysis of MDR isolates of K. pneumoniae detected during the monitoring of resistant Gram-negative bacteria at the National Medical Research Center for Children’s Health in 2014–2021.
Materials and methods. Whole-genome sequencing with a subsequent bioinformatics analysis of eight MDR isolates from the bloodstream and cerebrospinal fluid.
Results. MDR isolates belonged to 4 sublineages (SL): SL307, SL395, SL29 and SL1198. In the genomes of 6 pangrug-resistant (PDR) isolates, genes associated with resistance to all categories of antibiotics recommended for Enterobacteriaceae therapy were identified. Plasmids were present in all genomes. In 6 isolates, plasmids contained heavy metal ion resistance operons in addition to antibiotic resistance genes. Prophages within the plasmids were also involved in the transfer of resistance genes. The ST395 isolate from the cerebrospinal fluid belonged to the convergent pathotype in terms of resistance and virulence. Comparison of genomes within SLs revealed recombination events in the K- and O-locus regions and the Yersiniabactin operon.
Conclusion. Thus, in a sample of resistant K. pneumoniae isolated from bloodstream and cerebrospinal fluid, 6 PDR isolates were detected, one of which belongs to the convergent pathotype ST395.
 399-409
399-409



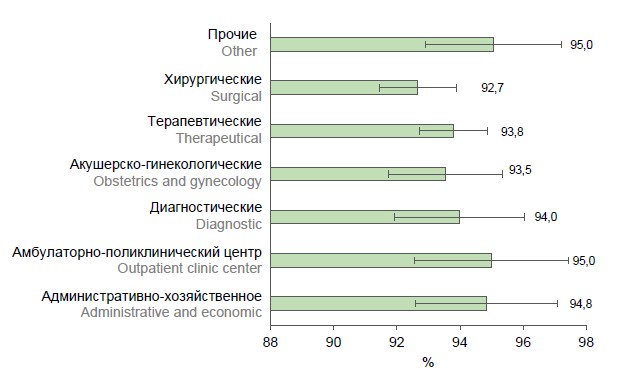
Results of screening for antibodies to varicella-zoster virus in healthcare workers of a multidisciplinary hospital in Moscow
Abstract
Introduction. Given the unfavorable epidemic situation with chickenpox and shingles in Russia, there is a high risk of virus introduction and spread in healthcare settings, including among medical staff who are not immune to varicella zoster virus (VZV).
The objective of this study is to assess the immunity of employees of a multidisciplinary hospital in Moscow to VZV.
Materials and methods. A selective screening study was carried out. Venous blood serum samples were taken from 1546 hospital employees as material for detection of IgG antibodies to VZV antigens using a commercial solid-phase enzyme immunoassay (ELISA) test system "Vecto VZV-IgG". All employees were questioned to obtain information about their infectious and vaccine history in relation to VZV.
Results and discussion. Screening for antibodies to VZV in the hospital workers revealed that 6.3% of those workers are not immune to VZV. The proportion of seronegative individuals was the highest (12.6 ± 2.4%) in the age group of 29 years and younger. VZV seronegative healthcare workers were found in various departments, but the presence of non-immune individuals among the staff of the obstetrics and gynecology departments (6.5%) is of epidemiologic concern. The results of the survey showed that documented data on infection and vaccination history cannot be used to assess the protection of healthcare workers against VZV infection.
Conclusion. The results of serologic screening for antibodies to VZV made it possible to identify a significant number of susceptible employees of the multidisciplinary hospital. In order to prevent the formation of multiple epidemic foci of varicella in medical organizations, it is advisable to include anti-VZV testing of medical staff in the state prevention programs with subsequent vaccination of non-immune individuals.
410-419
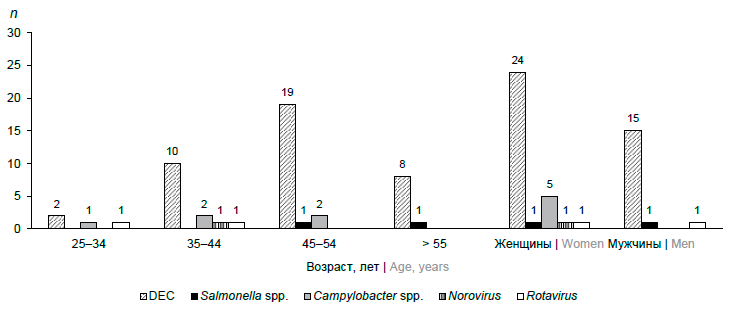
Molecular markers of acute intestinal infections in HIV-infected patients in the Chechen Republic
Abstract
Introduction. Diarrheal syndrome is the most characteristic symptom of HIV infection, which occurs in 70% of patients and is often fatal. The severity of diarrheal syndrome, irrespective of immune status, is usually determined by specific microorganisms colonising the gastrointestinal tract.
The objective of this study is to assess the prevalence of classical pathogens of acute intestinal infections in diarrheal syndrome in HIV-infected residents of the Chechen Republic (Grozny).
Materials and methods. Stool samples (n = 191) of HIV-infected patients with a history of diarrheal syndrome were studied by real-time PCR with two kits of reagents: "AmpliSens OKI screen-FL" for the detection of DNA/RNA of Shigella spp./EIEC, Salmonella spp., Campylobacter spp., Adenovirus, Rotavirus, Norovirus and Astrovirus; "AmpliSens Escherichiosis-FL" for the detection of diarrheagenic E. coli (DEC) DNA of five pathogroups: EPEC, EHEC, ETEC, EIEC, EAgEC.
Results. Genetic markers of the acute intestinal infection pathogens were detected in 20.9% of the examined individuals. In patients aged 0–7 years and 18–24 years, DNA/RNA of the tested pathogens were not detected. DNA of bacterial pathogens accounted for 93.9%, RNA of viral pathogens — 6.1%. The etiological structure of bacterial infections was represented by a significant predominance of DEC (84.8%) compared to 10.9% of Campylobacter spp. and 4.4% of Salmonella spp. The structure of viral infections included 66.7% Rotavirus and 33.3% Norovirus. Genetic markers of Adenovirus and Astrovirus have not been identified. In 77.5% of HIV-infected patients, diarrheal syndrome was caused by one pathogen (mono-infection), but in nine examined patients (22.5%) it had a combined etiology.
Conclusion. The etiology of acute intestinal infections in HIV-infected patients of the Chechen Republic includes bacterial and viral pathogens, in every fifth the cause of diarrheal disease was DEC. Due to diarrhea in HIV-infected people being a polyetiological disease, it is necessary to introduce a comprehensive, fast, reliable, and affordable method for identifying a wide range of pathogens that cause secondary infections.
420-427
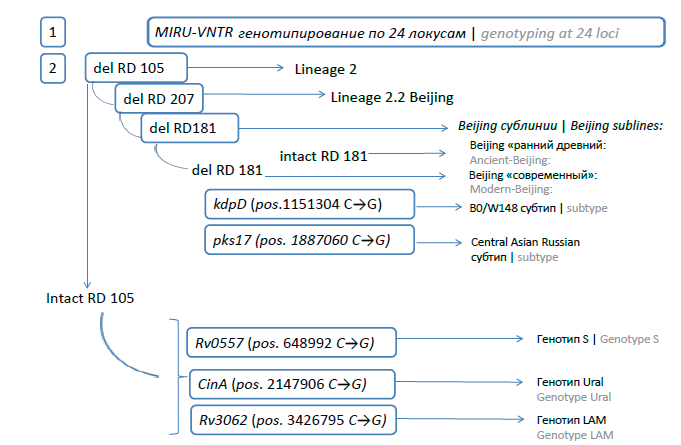
Genetic structure and drug resistance of Mycobacterium tuberculosis strains in the Kemerovo Region — Kuzbass
Abstract
Background. Kemerovo Region has a high burden of tuberculosis (TB) with incidence rates twice the national average. The circulating variants of Mycobacterium tuberculosis significantly influence the TB epidemic process. Screening of epidemically significant variants of the pathogen in areas with a high burden of TB underlies epidemiological diagnosis and is necessary for the development of effective prevention measures. However, the population structure of M. tuberculosis in the Kemerovo Region — Kuzbass is poorly understood.
Aims: to study genetic heterogeneity and phenotypic resistance to anti-tuberculosis drugs of M. tuberculosis strains in the Kemerovo Region.
Materials and methods. The MIRU-VNTR genotyping of 163 M. tuberculosis strains isolated from TB patients in the Kemerovo Region in March–October 2022 was carried out. Cultivation of M. tuberculosis, drug susceptibility testing, and isolation of genomic DNA were carried out by standard methods. Genotypic identification was performed using MIRU-VNTR (24 loci) typing. In parallel, express genotyping was carried out: identification of isolates of the Beijing genotype (by RD105/207) and non-Beijing; subtyping Beijing using real-time PCR tests for detection of Central Asian Russian and B0/W148; identification of the non-Beijing group by real-tine PCR RT tests for LAM, S, Ural.
Results. The isolates of the Beijing genotype (67.5%) were found to dominate both among newly diagnosed (64.4%) and previously treated patients (88.5%). MIRU-VNTR typing revealed 75 profiles, of which 94-32 (35.3%) and 100-32 (15.7%) were the most abundant and belonged to the Beijing genotype. Overall, 39.9% and 20.9% of isolates, respectively, were assigned to the Beijing Central Asian Russian and B0/W148 epidemic clusters, which differed significantly in MDR levels (50.8% and 85.3%, respectively; p = 0.005). The second most common were strains of the genetic family of the Euro-American lineage (L4) (31.9%): LAM (6.7%) Ural (7.4%) Haarlem (4.9%) and L4-unclassified (12.9%), MDR among of these minor genotypes was significantly lower than among Beijing genotype strains, and amounted to 11.5% (p < 0.001). Strains from HIV-TB patients (56.4% of the total sample) carried an MDR profile more often (54.8%) compared to TB cases without HIV infection (35.2%) (p = 0.005), which may be due to higher proportion of Beijing genotype strains in the HIV-TB group (75.0% vs. 57.7%; p = 0.026). Complete comparability of the SNP analysis (in-house tests) to identify the main genotypes and epidemically significant Beijing subtypes was shown, which made it possible to characterize 75.5% of the sample by the express method.
Conclusions. The molecular genetic screening carried out in the Kemerovo Region revealed the heterogeneity of the M. tuberculosis population, which was dominated by strains of the Beijing genotype, with the frequency of subtypes comparable with other territories of the Siberian Federal District.
428-441
The adaptive potential of North American subtype H7N2 avian influenza viruses to mammals
Abstract
Introduction. H7 subtype avian influenza viruses causing severe epizootics among birds are phylogenetically different in the Eastern and Western hemispheres. Numerous human infections caused by these viruses in the Eastern hemisphere indicate that H7 viruses can overcome the interspecies barrier and pose a potential threat of a new pandemic.The H7N2 viruses with deletion of amino acids 221–228 (H3 numbering) in hemagglutinin (HA) had been circulating among poultry in the Western Hemisphere during 1996–2006, and had once again been detected in 2016 in an animal shelter, where they caused cat diseases.
The objective of this study is to elucidate the mechanism of adaptation to mammals of North American H7N2 influenza viruses with deletion in HA.
Materials and methods. The A/chicken/New Jersey/294598-12/2004 (H7N2) virus was adapted to mice by the lung passages. Complete genomes of original and mouse-adapted viruses were analyzed. The receptor specificity and thermostability of viruses, HA activation pH and virulence for mice were determined.
Results. The non-pathogenic H7N2 avian influenza virus became pathogenic after 10 passages in mice. Amino acid substitutions occurred in five viral proteins: one in PB2 (E627K), NA (K127N), NEP (E14Q), four in HA and six in NS1. Mutations in HA slightly changed receptor specificity but increased the pH of HA activation by 0.4 units. The NS1 protein undergone the greatest changes in the positions (N73T, S114G, K118R, G171A, F214L and G224R), where amino acid polymorphisms were observed in the original virus, but only minor amino acid variants have been preserved in the mouse adapted variant.
Conclusion. The results show that H7N2 viruses have the potential to adapt to mammals. The increase in virulence is most likely due to the adaptive E627K mutation in PB2 and possibly in HA.
442-453
The increase in the incidence of syphilis in the Russian Federation: foreign migrant citizens as a risk group for the spread of the disease
Abstract
Relevance. The incidence of sexually transmitted infections (STIs) has a serious impact on the health and lives of children, adolescents and adults. Syphilis, like most STIs, is a socially significant disease, while among the factors influencing the spread of this infection, migration processes, including labor migration, occupy a special place.
Aims. To study syphilis in foreign migrants in the Russian Federation and individual subjects of the state in recent years.
Material and methods. A retrospective epidemiological analysis of the incidence of syphilis among the population of the Russian Federation and foreign migrants was performed. The data from Federal Statistical Monitoring Form No. 9 and No. 34 was used with reference to STIs incidence in 2011–2022 in Russia and in its regions.
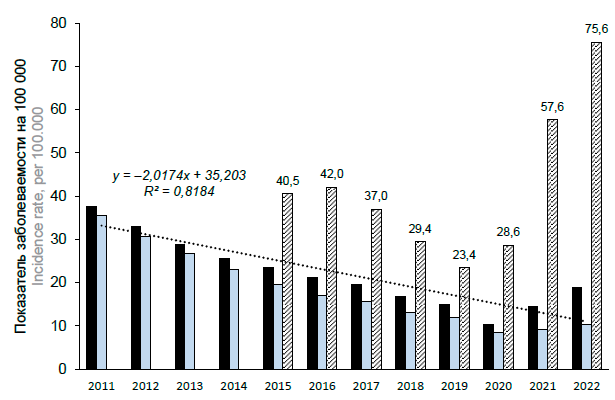
Results. Among foreign migrant citizens, the incidence rates were higher than the Russian average: 1.5–2.0 times before 2020, and 4 times in 2021 and 2022. The proportion of registered cases of syphilis among foreign citizens in 2021 was 36.4%, significantly exceeding this figure not only in 2020 (18.2%), but also in previous years (16.7–21.2%). An increase in the number of foreign citizens with syphilis was observed in 2021 in all federal districts of the Russian Federation, while the number of syphilis cases detected in this contingent of people varied significantly between different regions of the Russian Federation. The main share (98.1%) in the structure of syphilis detected in foreign citizens was latent forms of the disease.
Conclusions. The high level of detection of syphilis in foreign migrant citizens and the predominance of latent forms of the disease in this population represent a potential epidemiological danger of the spread of infection. In connection with the current situation, it seems necessary to develop and implement permanent and controlled algorithms for monitoring STIs in risk groups, including foreign migrant citizens.
454-461
Development of a methodology for molecular genetic monitoring for the cholera causative agent

Abstract
Background. The ongoing cholera pandemic determines the relevance of the development and improvement of methods for analysis of data on genome-wide sequencing of the cholera pathogen. This is of particular importance in the light of the challenges of import substitution of foreign products, including software.
The aim of the study was to develop a methodology for molecular genetic monitoring for the cholera causative agent using online geographic information system (GIS) and analysis with its help of strains isolated in Russia earlier.
Materials and methods. Data from genome-wide sequencing of 2598 toxigenic (ctxAB+tcpA+) strains of V. cholerae O1 El Tor, both obtained by the authors on the MiSeq (Illumina) platform, and retrieved from the NCBI database were used in the study. The SNP analysis software was developed in the Java and Python programming languages. Cytoscape program was used to visualize the dendrogram. The development of online GIS was carried out using the programming languages HTML, JavaScript and PHP. The freely distributed Leaflet library written in JavaScript was used as the core. Maps obtained from the OpenStreetMap community were used as cartographic data.
Results and discussion. A universal set of SNPs and software have been developed to analyze the data of genome-wide sequencing of cholera vibrio strains. It was shown that the majority of strains were distributed among several large clusters. The most closely related strains for cholera vibrions isolated in Russia since 2001 have been identified. An online GIS "Molecular genetic monitoring for V. cholerae" has been created, which allows the recognition of closely related strains directly on an electronic map.
462-471
Assessment of registered and hidden epidemic process of tick-borne encephalitis in the Republic of Karelia
Abstract
Introduction. The incidence of tick-borne encephalitis in the Russia remains relevant. The assessment of the epidemic process in the Republic of Karelia is important not only in terms of understanding its general patterns, but in connection with the growth of tourist attendance in the region.
Aims: To assess the current epidemic situation of tick-borne encephalitis in the Republic of Karelia, and to compare the characteristics of registered and hidden epidemic processes.
Materials and methods. The risk of infection and incidence were estimated based on the analysis of the registered cases of seeking medical help in connection with tick bites, the results of a study of tick-borne encephalitis virus (TBEV) infection rate in ticks, and the epidemiological investigation of cases of tick-borne encephalitis in 1992–2022. Clinical, gender and age structure, territorial distribution of patients and victims of the tick bites were compared with the results of serological studies of 2379 blood samples of the adult population, conducted in 2011–2022.
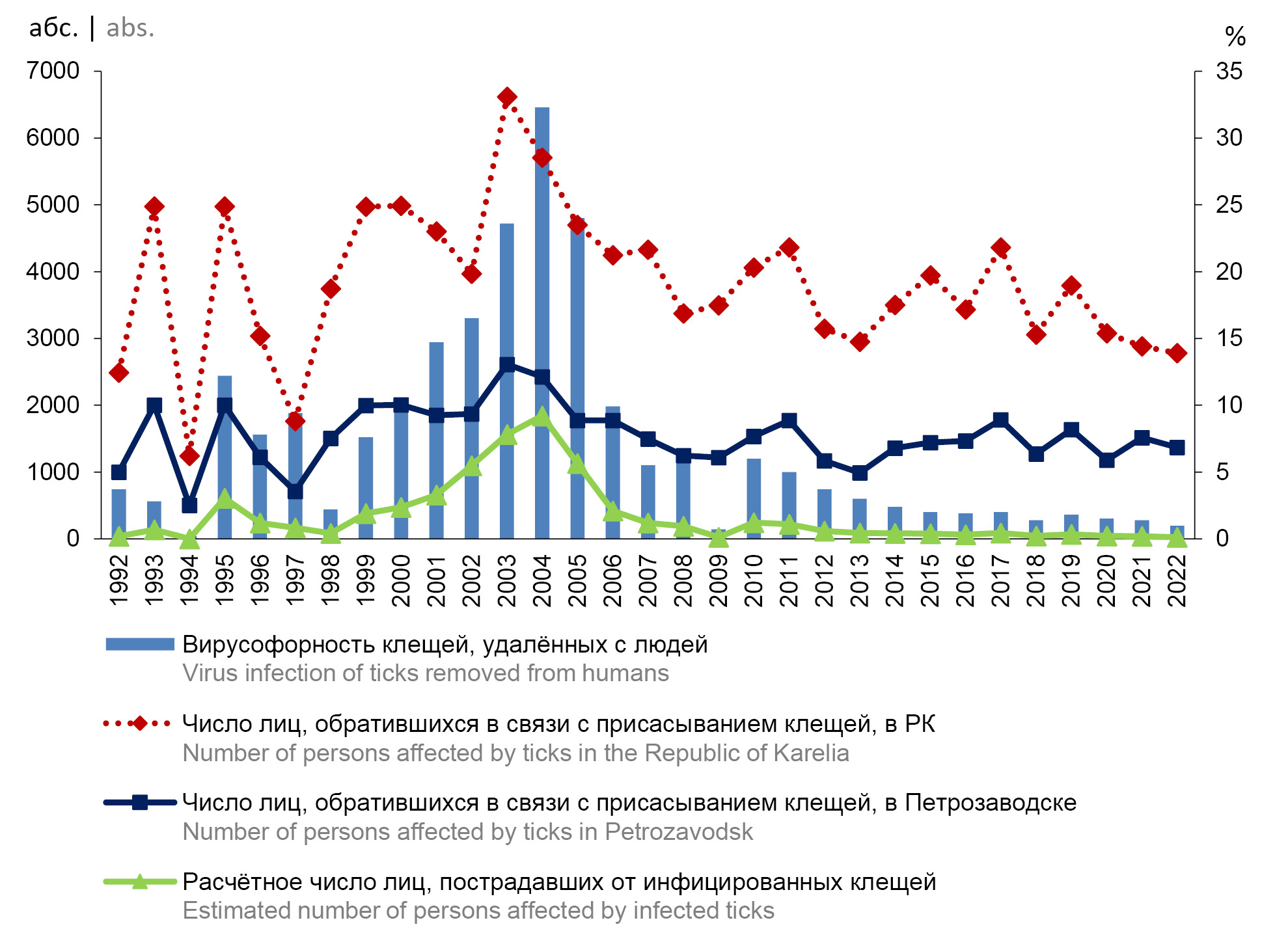
Results. Infection rates of ticks removed from humans ranged from 23.6–27.0% in 2002–2005 to 1.0% in 2022. In 2004, the TBEV antigen was detected in mosquitoes. The territory of risk is the southern part of Republic. However, the increase in number of cases of seeking medical help was observed in the northern part of Republic. The incidence rates exceeded the national average, especially in 2003–2004 (15.3–11.6 per 100 thousand). In 2021–2022, it decreased to 1.8–1.5 per 100,000. The dynamics of incidence had a high-degree correlation with the dynamics of seeking medical help and infection rates in ticks (R = 0.92 and 0.73). The reported incidence was lower than the estimated risk of infection. The meningeal forms of infection were most often diagnosed. The risk of the disease was higher in the elderly and in men, which was determined by the conditions of infection. Antibodies to TBEV were detected in 11.8 ± 0.7% of the examined persons.
Conclusion. A steady decrease in rates of registered tick-borne encephalitis incidence has been revealed in the Republic of Karelia, mainly due to the action of biological and natural factors. The assessment of seroprevalence made it possible to reveal the hidden part of the epidemic process.
472-484
Bacteria of genus Filifactor in patients with periodontitis and type 2 diabetes in accordance with metagenomic analysis of the periodontal microbiome
Abstract
Introduction. Periodontal diseases are a common pathology with chronic periodontitis (CP) being the most severe form. This polymicrobial disease has become a problem of great importance in recent years due to the possibility of development of systemic effects associated with this condition. CP is often combined with type 2 diabetes (T2D). The main cause of the occurrence and development of periodontal pathology is played by the bacteria Filifactor alocis, the least studied and most recently discovered periodontal pathogen.
The objective of this study was to identify bacteria of genus Filifactor as part of the periodontal microbiome associated with CP and T2D and to clarify the mechanisms of their possible influence on associated metabolic processes according to comparative metagenomic analysis.
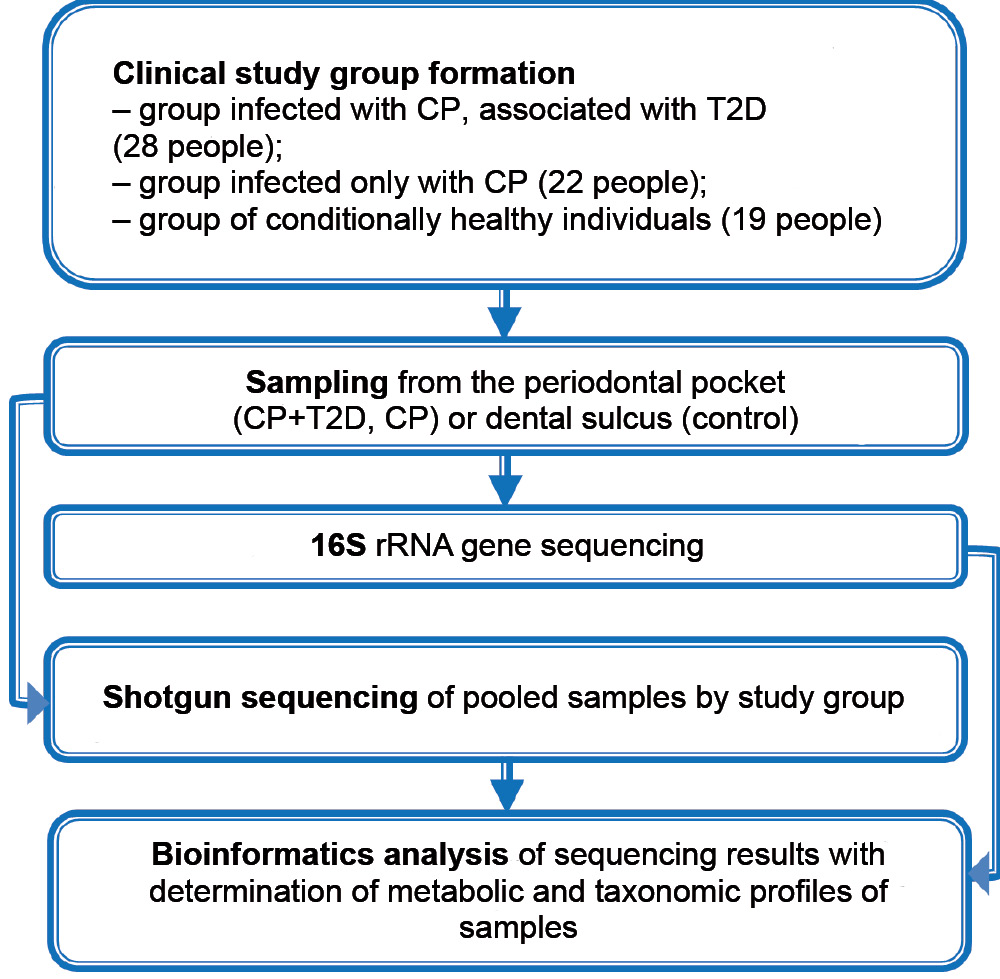
Materials and methods. A metagenomic study of the microbiome of periodontal pocket samples from 28 patients with CP associated with T2D and 22 patients with CP, as well as the microbiome of dental gingival sulcus samples from 19 clinically healthy individuals was performed. 16S-sequencing of the ribosomal RNA gene was used to determine the taxonomic composition of the microbiome. Prediction of metabolic pathways involving the microbiome was performed with the help of the shotgun method.
Results. Filifactor bacteria were the one of the most frequent microorganisms only in patients with CP associated with T2D. The rate of identification of these bacteria was correlated with low predicted metagenomic levels of fatty acid biosynthesis and pyrimidine metabolism in the affected area.
Conclusion. The detection frequency of Filifactor bacteria in patients associated with CP and T2D is negatively correlated with the selected features of putative metabolic pathways of the microbiome, which include fatty acid biosynthesis and pyrimidine metabolism.
485-494
REVIEWS
Accumulated experience and future prospects of in vivo hepatitis B virus research
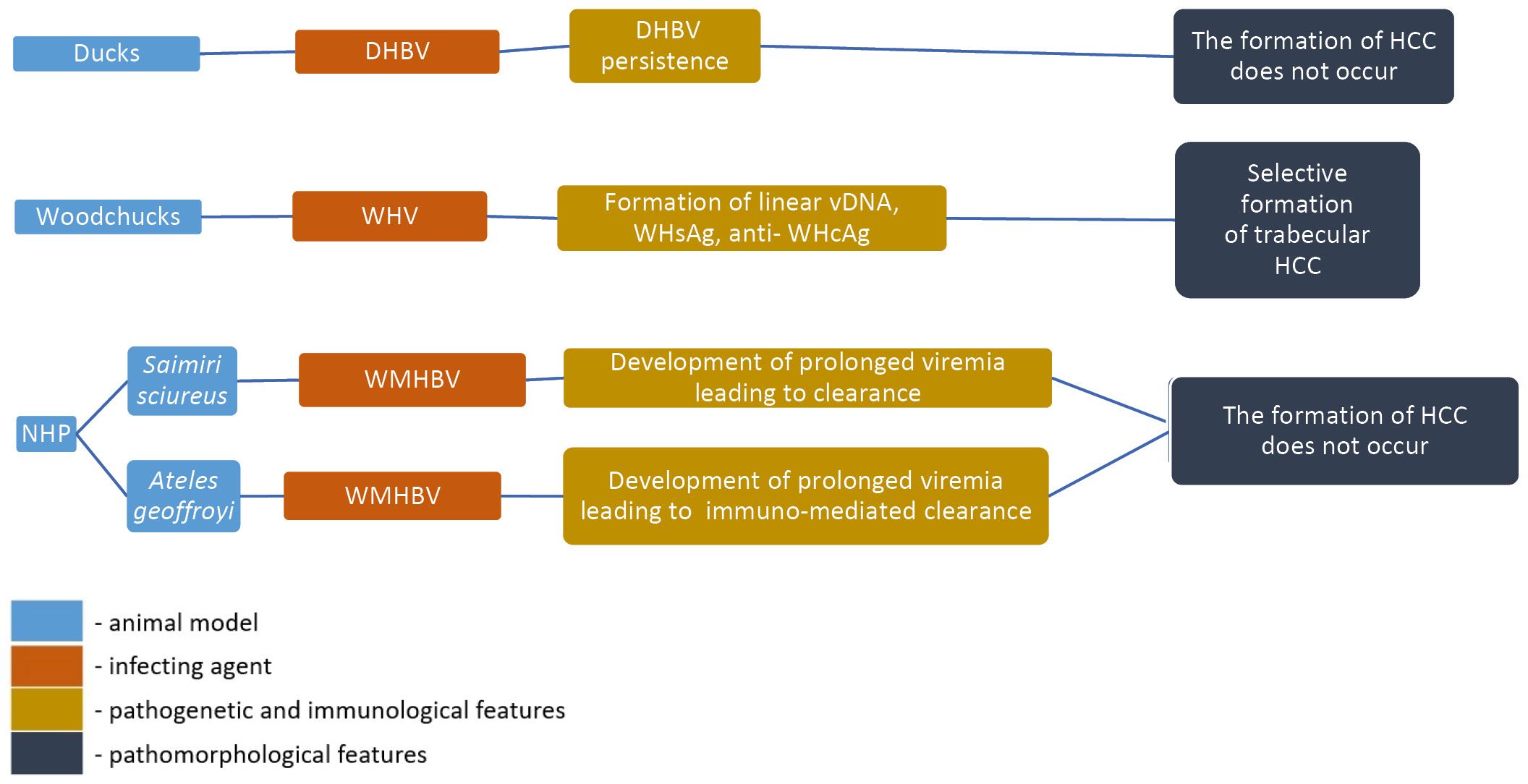
Abstract
Nowadays, an estimated more than 300 million people live with hepatitis B virus (HBV) infection globally. One of the main goals of the World Health Organization (WHO) is to eliminate viral hepatitis by the year 2030. The study of the pathogenic and immunologic properties of HBV, as well as therapeutic substances and treatment regimens, is significantly complicated by the insufficient number of susceptible biological test subjects (animal models) and the lack of zoonotic reservoirs of the virus. In this regard, researching the properties of HBV and related hepadnaviruses provides invaluable material for understanding the biology of the pathogen and the developing methods of prevention and control of this chronic infectious disease, leading to severe hepatopathies (cirrhosis and hepatocellular carcinoma).
Furthermore, prolonged HBV viremia leads to depletion of the immune system, reducing resistance against pathogens of other infections, especially those with a chronic course and socially determined spread.
The aim of this research is to evaluate existing animal models of HBV infection in the context of pathogenesis, immunologic and pathomorphological features. For the first time, the hypothesis of the possible use of certain models for the research of HBV-associated socially significant infections is considered from the point of view of the development of pathomorphological features.
To complete this review, we analyzed the information about the features of HBV infection models in vivo, published over the last 25 years in open sources (Web of Science, PubMed, Scopus, ScienceDirect, Springer). The main criteria for literature selection were the type of infecting agent, the observed immunologic features of the course of the infectious process and the availability of a description of the pathomorphological features in model organisms.
495-510
CHRONICLE
Results of the X Russian Scientific Conference with the participation of MSM “Persistence and symbiosis of events”
 511-512
511-512
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.