


Vol 101, No 3 (2024)

- Year: 2024
- Published: 16.07.2024
- Articles: 12
- URL: https://microbiol.crie.ru/jour/issue/view/184

ORIGINAL RESEARCHES
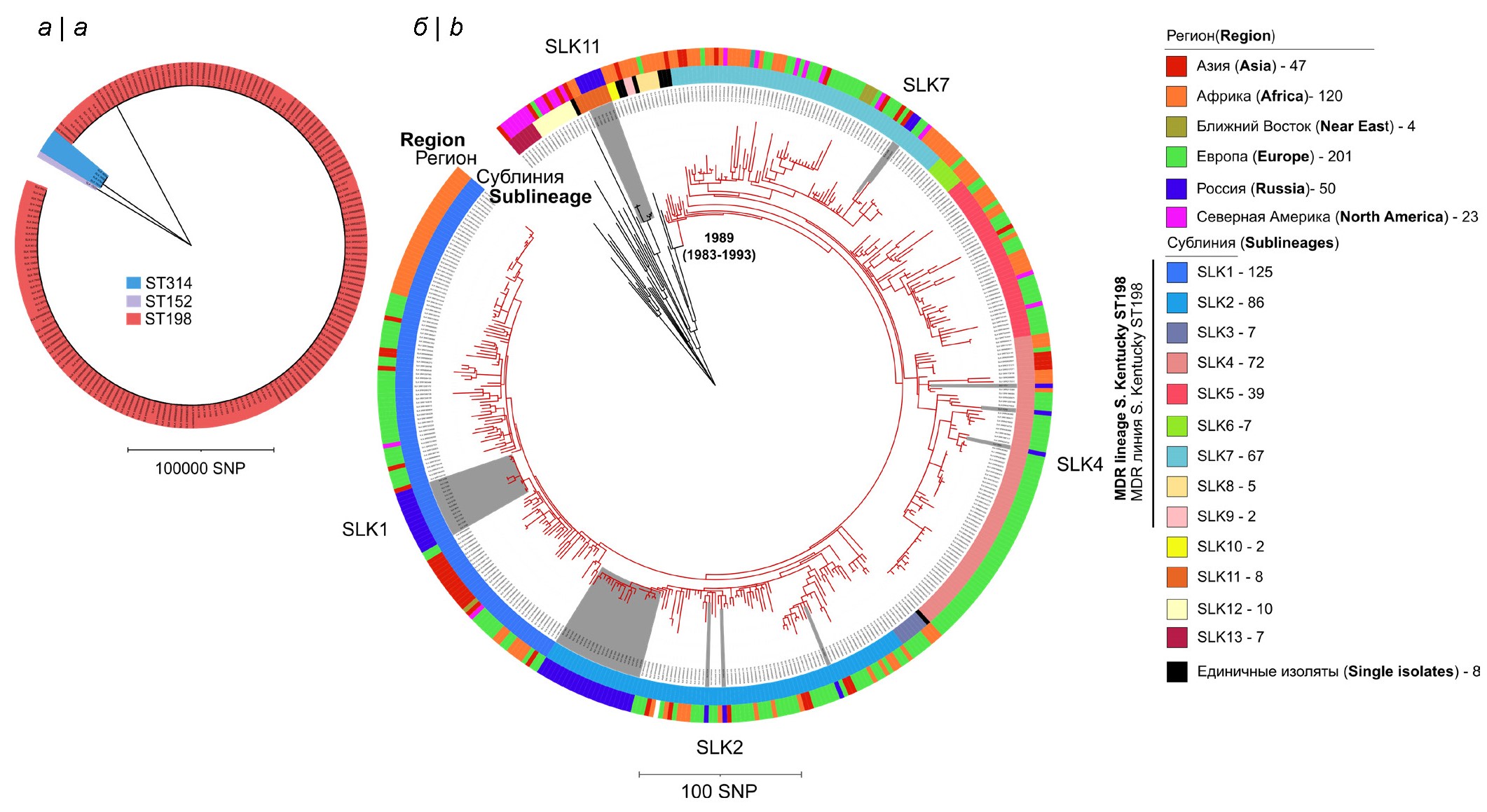
Genomic diversity and analysis of resistance determinants of Salmonella enterica subspecies enterica serotype Kentucky isolated in Russia
Abstract
Introduction. Salmonella Kentucky sequence type ST198 is one of the epidemiologically significant non-typhoidal Salmonella clones worldwide and is characterized by the presence of highly resistant strains and the ability to adapt to different animal hosts and environmental conditions.
The aim of this study was to analyze S. Kentucky strains isolated from various sources in Russia in terms of their phylogenetic position within the global diversity of the pathogen and their genetic characteristics.
Materials and methods. We examined 55 strains of S. Kentucky by whole-genome sequencing, which were isolated from 2010 to 2022 from various sources (clinical strains, food, as well as from farm animals, feed and environmental samples). Whole genome sequencing was performed using Illumina platforms. Phylogenetic analysis based on nucleotide variation analysis included an additional 390 S. Kentucky strains.
Results. Most of the Russian strains (n = 50) belonged to the ST198 sequence type, four strains were ST314 and one strain was ST152. Of the 50 Russian sequence-type ST198 strains, 44 belonged to the international monophyletic MDR lineage S. Kentucky ST198, and belonged to four separate sublineages, six strains occupying a basal position in relation to the MDR lineage. A total of 320 genes and mutations responsible for resistance to antimicrobial agents were identified. The most common were point mutations in the QRDR region. In most cases, Russian strains were characterized by the presence of variants of the SGI1-K genomic island. Moreover, the putative structure of SGI1 was correlated with the phylogenetic clustering of S. Kentucky sublineages.
Conclusions. The results of the study made it possible to assess the population structure of Russian S. Kentucky ST198 strains on a global scale and determine the genetic determinants of antibiotic resistance, including the structure of the SGI1 genomic island.
 303-314
303-314



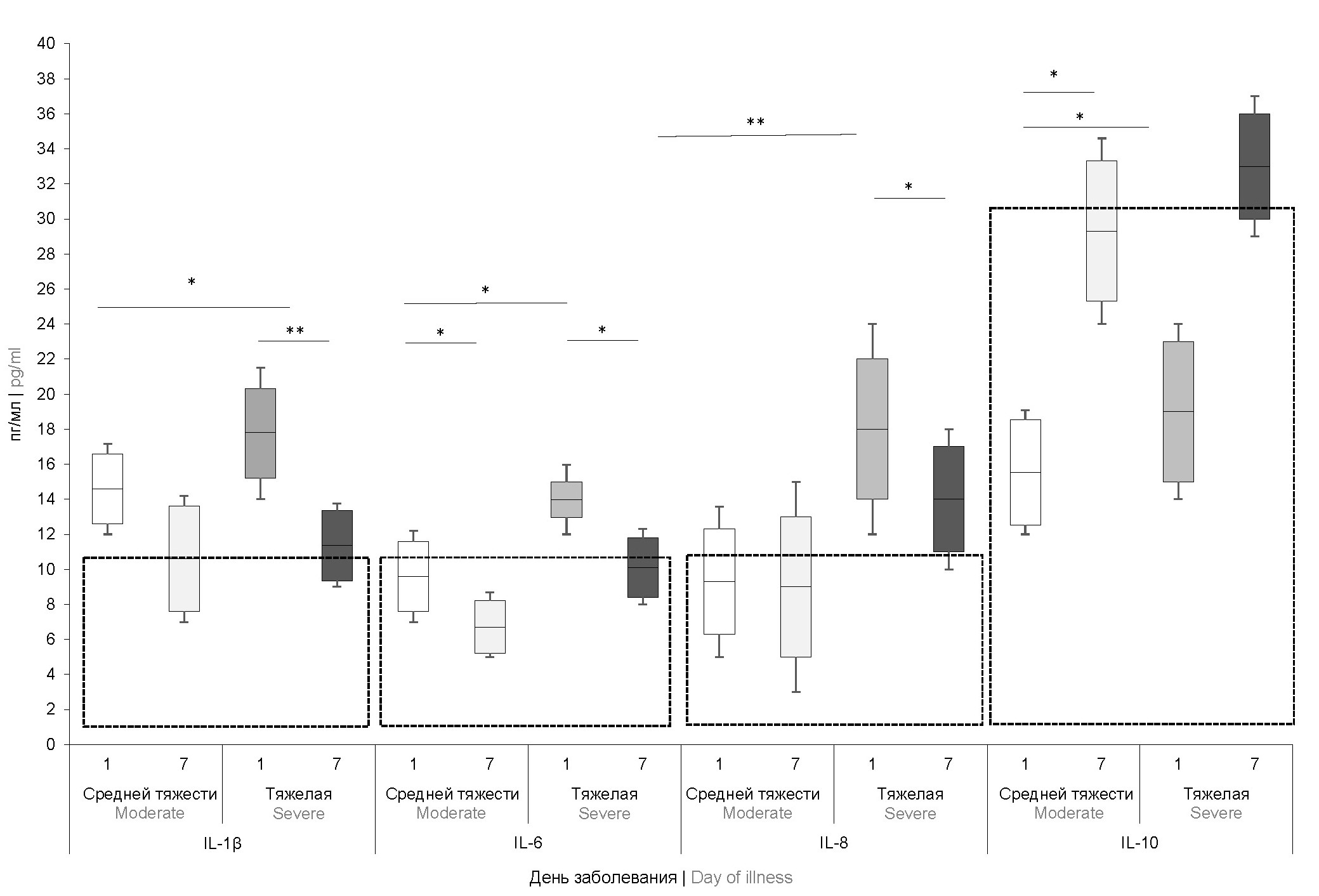
Campylobacteriosis: genotypic characteristics of the pathogen and immunological status of patients
Abstract
Introduction. Campylobacteriosis is among the leading causes of acute gastrointestinal infections. The severity of campylobacteriosis and the development of long-term complications may be influenced by the genotype of the pathogen, whose biological properties can affect immune response parameters.
The aim of the study was to identify common genotypes of epidemic clones of Campylobacter pathogens and to investigate characteristics of the immune response and severity of the disease.
Materials and methods. The study included 203 patients aged from 1 month to 17 years with campylobacteriosis who underwent treatment at the clinic of the Federal State Budgetary Institution "DNKCIB FMBA" in 2019–2021. The diagnosis was confirmed using polymerase chain reaction method. Patient samples were also analyzed using culture-based methods. Total DNA was extracted using the QIAamp DNA Mini Kit. Genetic determinants encoding virulence factors and MLST typing were performed using the ResFinder program. The immune status of patients was assessed on days 1 and 7 of the illness. Immunological investigation included measurement of serum immunoglobulin concentrations (IgA, IgM, IgG), C-reactive protein, and cytokines (IL-1β, IL-1, IL-2, IL-4, IL-5, IL-6, IL- 7, IL-8, IL-10, TNF-α, and IFN-γ).
Results. When analyzing the frequency of detection of Campylobacter sequence types in children with clinical intestinal infections, it was found that the profile of isolated isolates is most similar to those from countries of North America (USA and Canada), Northern Europe (Great Britain, Holland ) and Scandinavia (Denmark, Sweden, Finland). Identification of a pathogen with the flgE+, cdtA+, cdtC+ genotype was accompanied by a statistically significant increase in the level of IL-8 and a decrease in the content of IgA in the peripheral blood serum, which reflected the low efficiency of the immune response during infection with Campylobacter and predetermined the severe course of the infectious process during the disease.
315-326
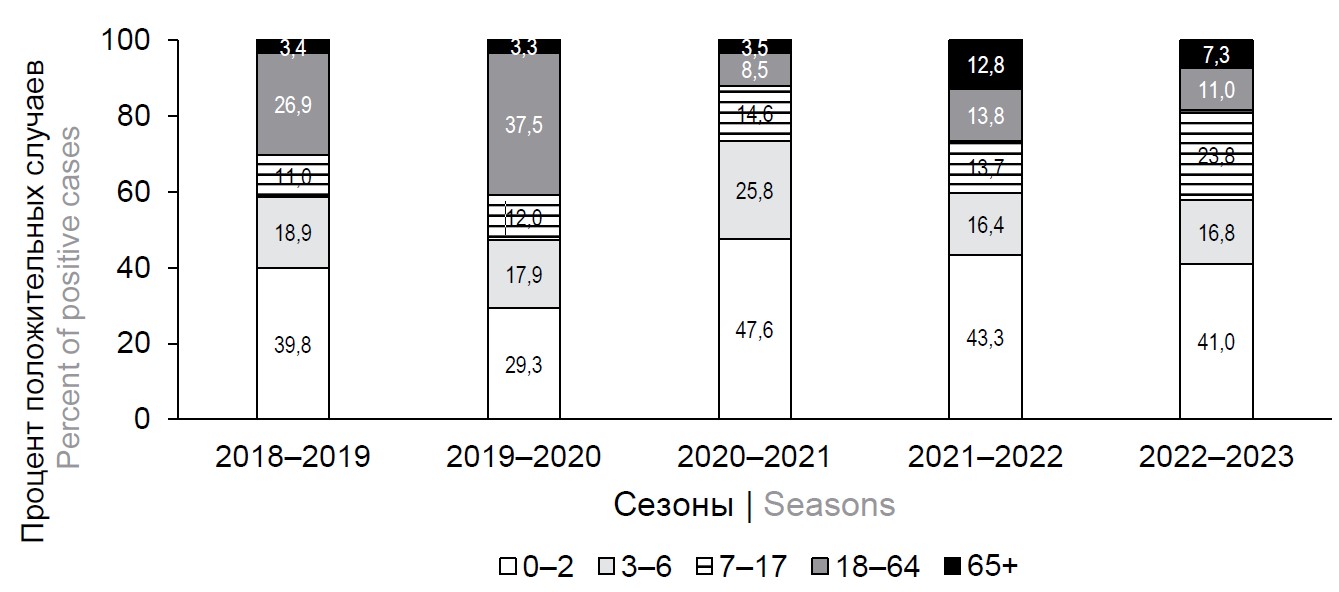
Changes in the etiological structure of severe acute respiratory viral infections in children and adults under the influence of the COVID-19 pandemic
Abstract
Introduction. The traditional surveillance system for influenza and ARVI provides a general description of epidemics, but does not provide information on the age-related characteristics of the etiology and clinical peculiarities of severe acute respiratory diseases (SARI) in hospitalized patients.
Aim. To monitor the etiology of SARI in hospitalized children and adults, assessing the impact of the COVID-19 pandemic on this process.
Materials and methods. Standardized clinical and laboratory monitoring of SARI among 18,458 hospitalized patients was carried out in hospitals in three cities of Russia with weekly PCR detection of 11 types of pathogens.
Results. According to the investigation of hospitalized patients with SARI for the period from 2018 to 2023, the viral etiology of respiratory diseases was deciphered in 58.3% of cases. Weekly monitoring showed a change in the etiological mosaic of SARI pathogens during the SARS-CoV-2 pandemic with a sharp decrease in the frequency of detection of influenza and respiratory syncytial virus (RSV) during the 2020–2021 season against the background of a significant increase of metapneumovirus and rhinovirus infections in children. During the 2022-2023 season an increase in the proportion of RSV infection in children under 6 years of age (up to 36.2%) was noted against the background of a significant decrease in the frequency of SARS-CoV-2. In the intensive care units (ICU), RSV infection was most often in children during the post-pandemic period (up to 30.1–53.6% of positive cases, p < 0.001); in adults, SARS-CoV-2 was mostly detected (76,5–100% of cases, p < 0.001).
Conclusion. Hospital surveillance data significantly complements the epidemiological information obtained in the traditional surveillance system. Monitoring of infections has shown a continuously changing etiological infrastructure of SARI, with the disappearance of influenza and RSV during the COVID-19 pandemic and their return to circulation in the post-pandemic period.
327-341
Molecular characteristics of fluoroquinolone-resistant Mycobacterium tuberculosis strains from newly diagnosed tuberculosis patients in the Northwest of Russia
Abstract
Introduction. Fluoroquinolones remain the key second-line anti-tuberculosis drugs.
The aim of the study was the molecular characterization of fluoroquinolone-resistant Mycobacterium tuberculosis strains from newly diagnosed tuberculosis patients in the Northwest of the Russian Federation.
Materials and methods. The retrospective study collection included M. tuberculosis isolates isolated in 2015–2019 from previously untreated tuberculosis patients. Susceptibility to antituberculosis drugs (including the fluoroquinolone ofloxacin) was determined using the BACTEC MGIT960 or absolute concentration method. Mutations in the gyrA gene as a marker of resistance to fluoroquinolones, were detected by real-time PCR. Beijing genotype and its subtypes were detected by PCR and real-time PCR methods. Non-Beijing strains were spoligotyped.
Results and discussion. Phenotypic resistance to ofloxacin was detected in 6.7% (40/599) of strains and in 17.4% (40/230) of MDR strains. 34 of 40 (85%) ofloxacin-resistant strains belonged to the Beijing genotype. 18 (45%) strains were assigned to the Russian epidemic subtype Beijing B0/W148 and 12 (30%) to Beijing Central Asian/Russian. The remaining 6 ofloxacin-resistant strains belonged to the Euro-American phylogenetic lineage. Mutations in the gyrA gene were found in 97.5% (39/40) of strains. The most common were mutations in codon 94 (69.2%, 27/39). The Asp94Gly substitution was identified in 57.5% (23/40) of ofloxacin-resistant strains and was dominant among Beijing (19/34) and non-Beijing (4/6) strains. The second most common substitution was Ala90Val (25%, 10/40). More than half of the ofloxacin-resistant strains, Beijing B0/W148 (10/18) and Central Asian/Russian (7/12), carried the Asp94Gly mutation.
Conclusion. In the Northwest of Russia in 2016-2019, primary resistance of M. tuberculosis to fluoroquinolones was 6.7% in the total collection and 17.4% of MDR strains, and was mainly caused by the gyrA Asp94Gly and Ala90Val mutations. Beijing B0/W148 genotype was characterized by the largest proportion of fluoroquinolone-resistant strains.
342-350
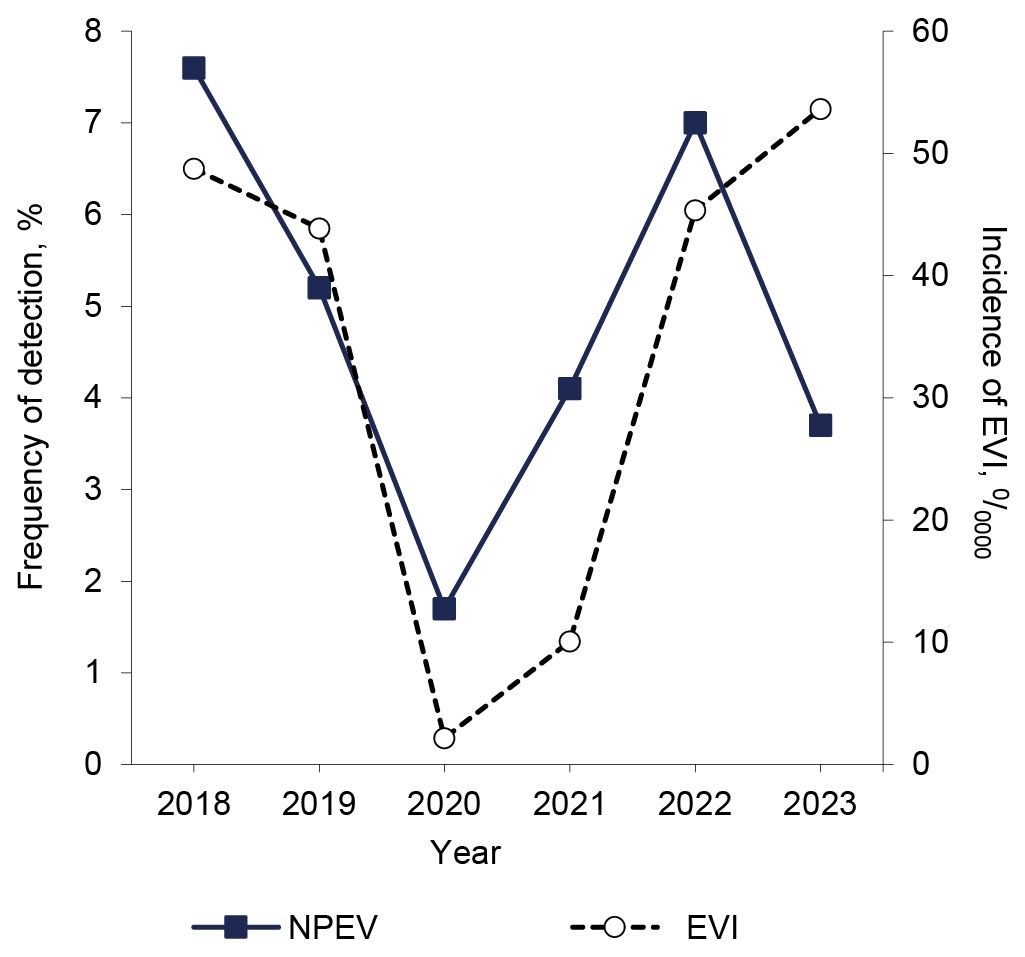
Detection and type identification of non-polio enteroviruses in children against the background of acute intestinal infections of various etiologies: 2018–2023
Abstract
Introduction. Enteroviruses (EV) are characterized by: species and type diversity, polymorphism of clinical manifestations, a tendency to epidemic spread, and are often the cause of disease outbreaks, which determines the relevance of monitoring EV strains in various clinical forms of infection, including in conditions of anti-epidemic measures.
The aim of the study: to characterize the prevalence and diversity of non-polio enteroviruses (NPEV) types in children with acute intestinal infection (AII) in the period 2018–2023, including the COVID-19 pandemic.
Materials and methods. The RT-PCR method was used to study 7302 samples of feces from children hospitalized with a diagnosis of AII in the infectious diseases hospital of Nizhny Novgorod. Genotyping of EV strains was carried out using fragment Sanger sequencing of the genome region encoding capsid protein 1 (VP1) and the online resource BLAST.
Results. EVs were found in 5.0 ± 0.3% (1.7–7.8%), both in mono- and mixed infections with other enteric viruses. The long-term dynamics of the frequency of EV detection and the incidence of EV infection in children in the Nizhny Novgorod region was characterized by a sharp decrease in indicators in 2020 against the backdrop of the introduction of anti-epidemic measures. When genotyping 299 strains, 41 types of NPEV of 4 species were identified. The spectrum included the main pathogens of exanthema and neuroinfections and rare types found in “minor” or intestinal forms of infection. During the study period, a redistribution of NPEV species was established. Before the pandemic, the ratio of Enterovirus A : Enterovirus B : Enterovirus C species was as follows — 41.0 : 46.7 : 12.3%; during the 2020 pandemic season the ratio was 0.0 : 37.5 : 62.5%; after the lifting of restrictive measures — 47 : 29 : 23%, which may be due to the different effectiveness of the restrictive measures on the mechanisms of transmission of EVs of different types.
Conclusion. The genetic diversity of NPEVs detected in children with AII complements information on the typical composition of the territorial enterovirus population. In children with AII, when the airborne transmission of SARS-CoV-2 was blocked, there was a decrease in the frequency of detection of viruses of the Enterovirus B type, the absence of detection of Enterovirus A and the constant presence of Enterovirus C.
351-361
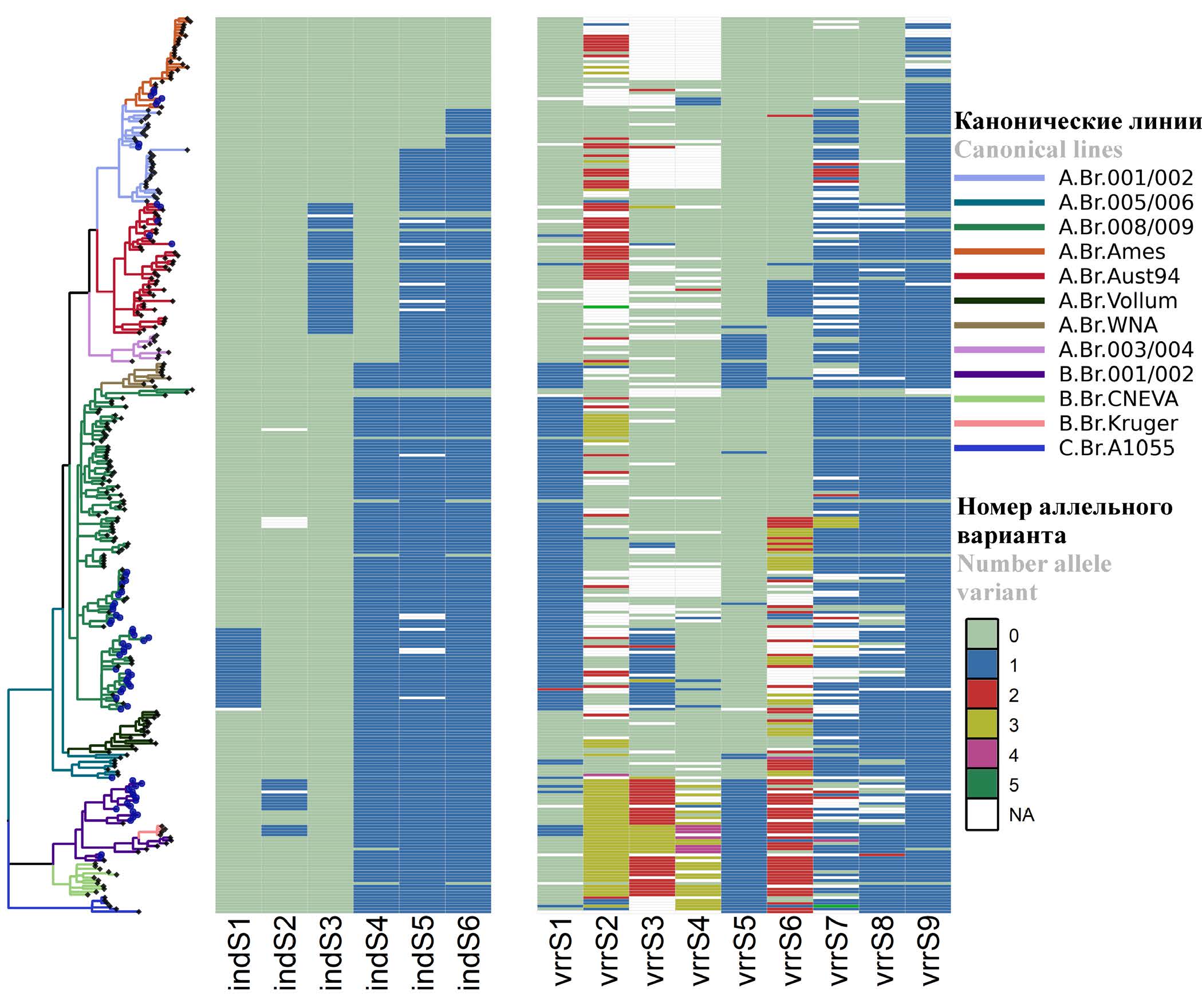
Development of a technique for molecular typing of Bacillus anthracis strains using new VNTR and INDEL markers
Abstract
Introduction. Bacillus anthracis, the pathogen of a particularly dangerous zoonotic disease known as anthrax, requires strict epidemiological control and is characterized by high genetic homogeneity, which necessitates the development of genotyping methods.
The aim of the study were to to find and characterize the VNTR and INDEL loci of B. anthracis and to develop on their basis a genotyping technique by PCR with electrophoretic detection of the results.
Materials and methods. Marker search and phylogenetic analysis were performed on a sample of 388 genomes of B. anthracis strains, 322 from the GenBank collection (RefSeq) and 66 from the collection of the Stavropol Anti-Plague Institute of Rospotrebnadzor. Phylogenetic analysis was performed on the basis of SNP crustal alignment using the Parsnp program. The search for markers was carried out using the Mauve program and author's scripts in Python. PCR was performed using a ScreenMix-HS kit (CJSC "Eurogen", Russia).
Results. Genomic variations of B. anthracis strains (SNP — 25,664, SNR — 14,387, VNTR — 693, INDEL — 14,667) were found, bioinformatic analysis of which revealed nine new VNTR and six INDEL molecular markers most suitable for genotyping. The genetic (allelic) variants of the markers are described. Primers were selected for the found markers and a PCR protocol with detection by electrophoresis in agarose gel was developed. When typing using VNTR markers was applied, the strains were divided into nine clusters: A.Br.Ames, A.Br.001/002, A.Br.Aust94, A.Br.005/006, A.Br.008/009 (Tsiankovskii), A.Br.008/009 (STI), A.Br.008/009 (A.Br.125), A.Br.008/009 (strain 228/269), B.Br.001/002. When typing using INDEL markers, the strains were divided into six clusters: A.Br.Ames, A.Br.001/002, A.Br.Aust94, A.Br.008/009(Tsiankovskii), B.Br.001/002(B.Br.014), as well as a cluster comprising several genetic lineages: A.Br.008/009 (STI), A.Br.008/009 (A.Br.125), A.Br.005/006 и B.Br.001/002.
Conclusion. The use of the developed methodology for the identification of variable VNTR and INDEL loci makes it possible to reliably determine the phylogenetic position of B. anthracis strains and is promising for use in the epidemiological investigation of anthrax outbreaks.
362-371
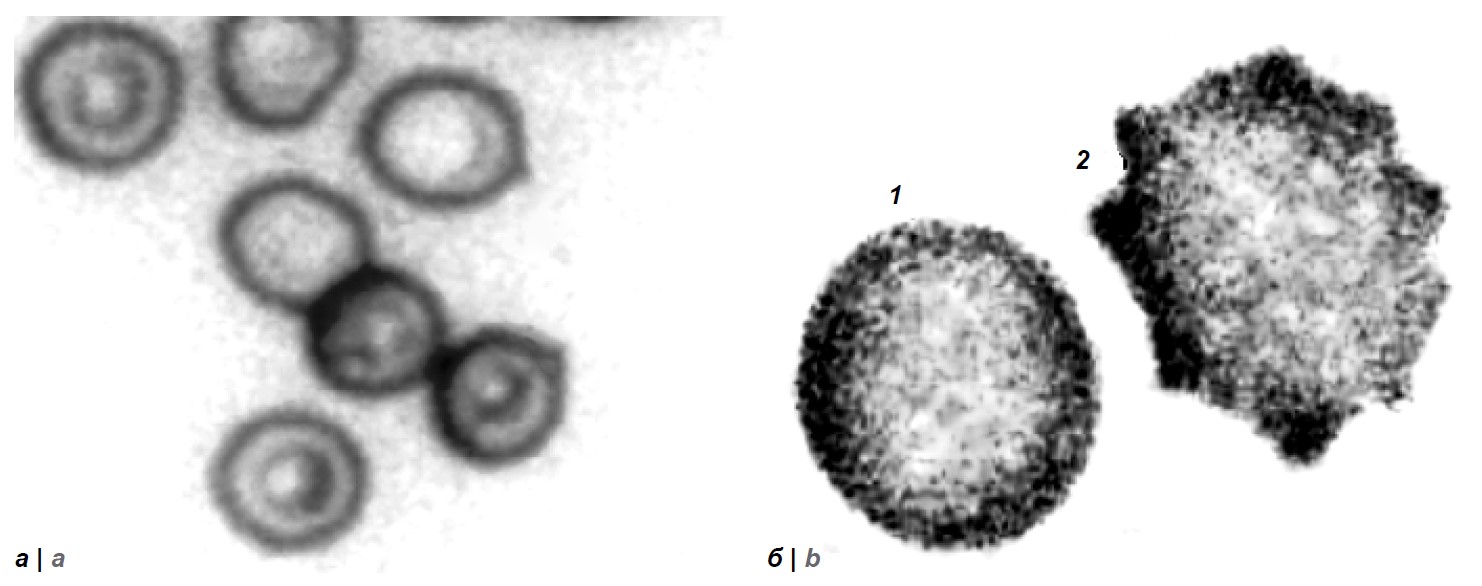
Influence of Yersinia pestis with different plasmid composition on the erythrocyte membrane in the blood of guinea pigs
Abstract
Introduction. Based on data on the role of blood erythrocytes in the development and implementation of the vaccine and infectious processes in plague, it was of interest to evaluate changes in erythrocyte surface architecture from the position of searching for informative criteria for the preclinical evaluation of anti-plague vaccines.
Aim — using atomic force microscopy to characterize the state of the blood erythrocyte membrane of guinea pigs in response to subcutaneous administration of the vaccine strain Yersinia pestis EV NIIEG and its isogenic derivatives.
Materials and methods. For immunization of animals strain Y. pestis EV NIIEG (pYT+, pYV+, pYP+) and its isogenic derivatives Y. pestis KM216 (pYT–, pYV–, pYP+), Y. pestis KM217 (pYT–, pYV+, pYP–), Y. pestis KM218 (pYT–, pYV–, pYP–) were used. Analysis of the erythrocyte membrane was carried out using a Solver P47-PRO scanning probe microscope (NT-MDT, Russia).
Results. The most pronounced changes in the surface architectonics of the membrane of guinea pig erythrocytes were established during the first three days of the formation of the immune response to the Y. pestis EV strain NIIEG and its isogenic variant Y. pestis KM217, in the genome of which the pYV plasmid is preserved, administered at a dose of 5 × 108 CFU. A significant increase (p < 0.05) in the proportion of transformed cell forms (43.67 ± 3.63% and 37.83 ± 7.03% versus 4.08 ± 0.86% in the control group), root mean square roughness (319 ± 8 nm and 312 ± 7 nm versus 70 ± 6 nm in the control group), Young's modulus (125.73 ± 4.48 kPa and 113.8 ± 5.41 kPa versus 53.03 ± 1.47 kPa in the control group). By the 21st day, the value of these indicators decreased by an average of 2.7, 2.0 and 1.5 times, respectively, indicating restoration of the erythrocyte membrane.
Conclusion. The dependence of the changes in the erythrocyte membrane and the rate of their restoration on the plasmid composition of Y. pestis strains has been established. The data obtained contribute to the understanding of the processes of interaction of Y. pestis with the erythrocyte membrane and can be used as additional characteristics in the development of new criteria for preclinical evaluation of plague candidate vaccines.
372-381
The effect of carbohydrate adjuvants in the composition of the experimental hantavirus vaccine on the dynamics of neutralizing antibodies in the blood sera of guinea pigs
Abstract
Introduction. Vaccination could be the most effective means of preventing hemorrhagic fever with renal syndrome (HFRS), which is one of the leading zoonoses and a major cause of natural disease in humans. Therefore, research to develop an effective vaccine and the search for new non-toxic and effective adjuvants that can enhance and prolong the immune response, reduce the antigen concentration and reduce the frequency of dose administration is an urgent task.
The aim of the study was to determine the dependence of the immunogenic activity of an experimental hantavirus vaccine based on Puumala virus (HV) on the antigen dose both in native form and in combination with carbohydrate adjuvants.
Materials and methods. The immunoadjuvant effect of Ac3-LPS S. sonnei and S. flexneri 1B and emulsions of nanoemulsified squalene Sepivac SWE (SWE) as part of HV at the optimal and minimal doses was investigated by induction of neutralising antibodies after 3-fold immunisation (day 0, 14, 182) of guinea pigs (Cavia porcellus). Differences in the immunogenic activity of HV were assessed in the neutralisation reaction by 50% suppression of focal units in Vero E6 cell cultures.
Results. A statistically significant increase in neutralising antibodies levels was observed after immunization both with HV at the optimal dose and for its combination with adjuvants. Ac3-LPS S. sonnei and SWE showed the most pronounced immunoadjuvant effect at concentrations of 50 and 100 μg/dose, respectively. The immunoadjuvant effect of the combination of the hantavirus vaccine with Ac3-LPS S. sonnei and SWE contributed to both an enhanced immune response and its duration. The tenfold reduction of the antigen dose in the presence of SWE allows optimization of the immune response to the vaccine.
Conclusion. The results of this study show the prospects of using Ac3-LPS S. sonnei and Sepivac SWE in the vaccine for the prevention of HFRS.
383-392
REVIEWS
Hospital environment microbiome
Abstract
The aim of the review is to give a brief description of the biodiversity and structure of the hospital environment microbiome based on molecular genetic research methods.
Until a certain time, studies of the hospital environment microbiota for the purposes of epidemiological surveillance and control of healthcare-associated infections (HAIs) were based on routine microbiological identification of clinically relevant bacterial taxa. Discovery of DNA, the development of sequencing technologies, PCR and cloning techniques enabled the investigation of microbial communities using cultivation-independent, DNA and RNA-based approaches. At the current level of knowledge, the hospital environment can be considered as a superorganism with its own microbiome. Multiomic technologies, including meta-transcriptomic, meta-proteomic and metabolomic approaches, provide detailed information about microbial activity in the environment. Now it has been established that there is a stable core of the hospital microbiome where the vast majority of microorganisms are necessary for the functioning of the hospital ecosystem and are not classified as human pathogens. The hospital microbiome has a homogeneous structure composed by a massive dominance of a few taxa and microbial network with low connectivity forming a clustered topology. A keystone species is a taxon whose importance for maintaining community structure is relatively higher than others and its identification is of paramount importance. Due to the lack of knowledge of the hospital environment microbiome by molecular genetic technologies, there is no single shared point of view on the microbial diversity in different healthcare facilities. But there is no doubt that molecular genetic technologies will shed light on the evolution of hospital strains and determine which indicators are the most informative for monitoring and prognosis of HAIs.
393-398
Analysis of aerobiological studies with orthopoxviruses by U.S. Department of Defense
Abstract
Discontinuation of vaccination after the completion of Smallpox global eradication program led to a sharp decrease in the level of collective immunity not only to smallpox but also to other orthopoxvirus infections. Over the past 10–15 years, the world has seen an increase in the frequency of diseases caused by smallpox viruses of cows, buffaloes, camels. The outbreak of mpox (a disease caused by the monkey pox virus) occurred in 2022–2023. Analysis of the literature data on the organization of the orthopoxvirus genome suggest that smallpox could have occurred in the past as a result of evolutionary changes in the zoonotic progenitor virus. In this regard, there is a threat of a new particularly dangerous anthropozoonosis, the pathogen of which can occur both naturally and artificially.
The aim of the review is to analyze open science published data on aerobiological research with OPVs conducted by the U.S. Department of Defense from 1994-2013, which was a period of restricted research and storage of smallpox virus samples. The authors did not find any publications of the results of aerobiological research with orthopoxviruses conducted by the US Department of Defense after 2013 in open scientific sources.
The review presents a data analysis in Russian and English-speaking scientist publication as well as those posted on the Internet.
The presented results of aerobiological studies with orthopoxviruses indicate the interest of the US military department in carrying out experimental work of dual use, including monitoring of the properties of orthopoxviruses and a possible change in their pathogenicity for humans, selection of optimal laboratory models for studying the properties of orthopoxviruses, and the possibility of modeling the properties of the smallpox virus when using other orthopoxviruses (cowpox virus, rabbit pox virus, monkey pox virus), modeling of the main characteristics of the disease caused by the smallpox virus in humans and evaluation of the effectiveness of existing and newly developed vaccines against smallpox, comparative study of effectiveness of antiviral drugs for regular or post-exposure prophylaxis of naturally occurring smallpox and monkey smallpox.
399-411
Monoclonal antibody techniques. 50 years of development
Abstract
Monoclonal antibodies are widely used in all fields of biology and medicine. The emergence and development of the technology for their production revolutionized immunology and allowed the creation of not only new diagnostic methods, but also many effective drugs. The purpose of our review is to analyze and summarize relevant data concerning the technology of obtaining monoclonal antibodies. We have analyzed information from 70 modern literary sources devoted to various methods of obtaining them. In this review, we tried to cover the entire range of methods used to obtain monoclonal antibodies today.
412-427
ANNIVERSARIES
On the occasion of the 70th anniversary of the Professor Murad Giyas oglu Mammadov
 428-428
428-428
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.