


Genomic diversity and analysis of resistance determinants of Salmonella enterica subspecies enterica serotype Kentucky isolated in Russia
- Authors: Kuleshov K.V.1, Pavlova A.S.1, Kremleva A.A.2, Karpenko A.E.1, Mikhaylova Y.V.1, Krutova N.E.1, Lisitsyna M.R.1, Popova K.R.1, Veselova O.A.1, Podkolzin A.T.1, Akimkin V.G.1
-
Affiliations:
- Central Research Institute for Epidemiology
- Federal Center for Animal Health
- Issue: Vol 101, No 3 (2024)
- Pages: 303-314
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18523
- DOI: https://doi.org/10.36233/0372-9311-488
- EDN: https://elibrary.ru/owlgtw
- ID: 18523

Cite item

Abstract
Introduction. Salmonella Kentucky sequence type ST198 is one of the epidemiologically significant non-typhoidal Salmonella clones worldwide and is characterized by the presence of highly resistant strains and the ability to adapt to different animal hosts and environmental conditions.
The aim of this study was to analyze S. Kentucky strains isolated from various sources in Russia in terms of their phylogenetic position within the global diversity of the pathogen and their genetic characteristics.
Materials and methods. We examined 55 strains of S. Kentucky by whole-genome sequencing, which were isolated from 2010 to 2022 from various sources (clinical strains, food, as well as from farm animals, feed and environmental samples). Whole genome sequencing was performed using Illumina platforms. Phylogenetic analysis based on nucleotide variation analysis included an additional 390 S. Kentucky strains.
Results. Most of the Russian strains (n = 50) belonged to the ST198 sequence type, four strains were ST314 and one strain was ST152. Of the 50 Russian sequence-type ST198 strains, 44 belonged to the international monophyletic MDR lineage S. Kentucky ST198, and belonged to four separate sublineages, six strains occupying a basal position in relation to the MDR lineage. A total of 320 genes and mutations responsible for resistance to antimicrobial agents were identified. The most common were point mutations in the QRDR region. In most cases, Russian strains were characterized by the presence of variants of the SGI1-K genomic island. Moreover, the putative structure of SGI1 was correlated with the phylogenetic clustering of S. Kentucky sublineages.
Conclusions. The results of the study made it possible to assess the population structure of Russian S. Kentucky ST198 strains on a global scale and determine the genetic determinants of antibiotic resistance, including the structure of the SGI1 genomic island.
Full Text
Introduction
One of the epidemiologically significant serotypes of nontyphoidal salmonellae is the Salmonella enterica subspecies enterica serotype Kentucky (hereinafter referred to as S. Kentucky). A number of studies have described cases of isolation of strains producing extended-spectrum β-lactamases (ESBLs), and in some cases even resistant to carbapenems. S. Kentucky is considered one of the target serotypes for monitoring prevalence in poultry production [1].
Various studies in recent years have shed light on the emergence, drivers, and potential threats of S. Kentucky, in particular the sequence type (ST) ST198, which readily adapts to the selection pressure exerted by antibiotic use under various environmental conditions. This Salmonella serotype has evolved from no resistance before 1990 to an increase in the prevalence of ciprofloxacin-resistant isolates in the early 21st century (from 55% in 2007 to 88% in 2017 [2, 3]). In the first years of this decade, studies record the accumulation of genetic elements responsible for resistance to a wide range of antimicrobial drugs [2]. The intensive use of antibiotics in animal husbandry and medicine is one of the primary causes of this phenomenon. Globalization of trade and travel opportunities create favorable conditions for the spread of resistant bacterial clones internationally. In Europe, human infection with the strain S. Kentucky ST198 multidrug-resistant (MDR) strain was previously mainly associated with travel to North Africa or Southeast Asia, with Egypt being the most likely source of these bacterial isolates [2, 4, 5].
It is shown that all MDR strains of S. Kentucky ST198 belong to a single genetic lineage that has accumulated antimicrobial resistance determinants since the early 1990s. [5]. The different determinants of chromosomal resistance since the mid-1990s are associated with Salmonella genomic island 1 (SGI1) integration. SGI1 is a 43,000 bp genomic island originally described in S. Typhimurium DT104 [6] and encodes resistance to a variety of antimicrobial drugs, including amoxicillin, gentamicin, and sulfonamides [7]. The insertion of SGI1 was followed by cumulative mutations in the gyrA and parC genes, leading to resistance to nalidixic acid and then to ciprofloxacin in 2002. It is assumed that it is due to the peculiarities of genetic organization — chromosome-integrated resistance – that explains the clonal success of this MDR lineage and its rapid and wide spread worldwide [5, 8]. Previous studies have demonstrated that the acquisition of SGI1 compared to the acquisition of resistance plasmids does not require the cost of adaptation (bacterial fitness) during growth under nutrient-limited conditions [9], which may directly affect the effective spread of antibiotic-resistant strains and makes it necessary to closely monitor the trends and dynamics of circulation of this pathogen not only regionally but also globally.
Existing studies based on whole genome sequencing data and population structure analysis have provided a useful basis for understanding the ongoing evolution of the MDR lineage of S. Kentucky ST198 [5]. MDR-lineage strains were mainly found in Egypt until 2005 and then rapidly spread throughout Africa and the Middle East [4]. Another cause for concern is the expanding range of sources of detection of MDR-lineage strains of S. Kentucky ST198. Initially, they were detected in autochthonous poultry, but later in various animals and food products (Salmonella-contaminated spices in France and the USA, turkey flocks in Germany and Poland, and wild animals) [4, 10–12].
According to salmonellosis monitoring data, S. Kentucky is not among the ten dominant serotypes of salmonellae in Russia. The cases of salmonella detection from various sources do not exceed 0.3% during the last year1, which corresponds to several dozens of strains per year. At the same time, according to published data, this serotype was not an etiologic agent of cases of group morbidity from 2019 to the present [13], and information about the isolation of strains of S. Kentucky ST198 in Russia is limited to single cases [14]. At the same time, the question of the population structure of Russian strains and their genetic characteristics in comparison with strains circulating in other regions of the world remains open.
The aim of this study is to analyze S. Kentucky strains isolated from various sources in Russia in terms of their phylogenetic position in the scale of global pathogen diversity and determination of their genetic features.
Materials and methods
Selection of isolates and microbiological studies
Between 2010 and 2022, 55 strains of S. Kentucky isolated on the territory of the regions of the Russian Federation from various sources were studied.
Microbiological analysis of Salmonella strains
Prior to total DNA extraction for whole genomic sequencing, bacterial culture was sieved to single colonies and serotype confirmation was performed using polyclonal (PETSAL) and monoclonal (Sifin) sera.
Whole-genome sequencing
Total DNA from 7 × 109 CFU was extracted using the Ribo Prep kit (AmpliSens). Genomic libraries for whole genome sequencing of each Salmonella strain were prepared from 70 ng of total DNA using the NexteraXT kit (Illumina). Massively parallel sequencing was performed on MiSeq, Hiseq, and NextSeq (Illumina) platforms.
Processing of whole genome sequencing data and de novo assembly of genomes
Removal of adapters and low quality nucleotide reads was performed using the BBTools package2. De novo assembly of contigs for each of the strains was performed using the SKESA assembler [15]. The number of nucleotide reads and assembly quality were considered sufficient for further analysis under the following conditions: (1) an average of more than 30-fold sequencing depth was achieved by reverse mapping the original nucleotide reads to the assembled contigs; (2) the requirements for the metrics of the obtained contigs were met3: total contig length from 4 to 5.8 million bp, N50 more than 20,000 bp, number of contigs less than 600, proportion of "N" nucleotides less than 3%, and percentage of contigs belonging to the Salmonella genus more than 70%. At the final stage of assembly validation, the obtained contigs were used to confirm the belonging of the sequenced strain to the serotype S. Kentucky using the SISTR [16] and SeqSero [17].
Phylogenetic analysis
The total collection of genomes for phylogenetic analysis included 445 genomes of S. Kentucky strains, including 55 strains of Russian origin (GenBank accession numbers SAMN42109132–SAMN42109186) and 390 strains sequenced in previous studies [5, 9, 18, 19]. Bioinformatic analysis was based on the search for informative single nucleotide polymorphisms (SNPs) by mapping the nucleotide reads of each of the sequenced bacterial isolates to the reference genome of S. Kentucky (NCBI accession number: CP028357) using the SnapperDB software pipeline [20]. The analysis was performed with the following parameters: minimum consensus depth — 10x, minimum read mapping quality to account for variation — 30x, minimum major variant fraction — 90%, minimum required genome-wide average coverage — 30x. The resulting SNP profiles were converted to alignment format. Subsequent phylogenetic tree reconstruction and removal of SNPs located in recombination regions were performed in the Gubbins v. 3.2.1 program [21]. Bootstrap analysis with 1000 repetitions was performed to confirm the tree topology. Phylogenetic sublineages (genetically homogeneous groups of SNP profiles) were determined using the Fastbaps program with the parameters "optimize.baps". As a result, the clusters identified at the first level of clustering were used [22].
Determination of sequencing type, genetic determinants of antibiotic resistance and plasmid replicons
ST based on 7 housekeeping genes were determined using mlst program4. Antibiotic resistance genes and point mutations were searched using AMRFinderPlus v. 3.10.40 with the parameters "-I 0.9 -c 0.6 -O Salmonella" (minimum percentage of identity — 90%, minimum overlap — 60% with filtering of results specific to the Salmonella genus) [23]. Plasmid type determinations were performed using the MOB-suite v. 3.0.0 program [24].
SGI1 analysis
We determined the presence and analyzed the putative structure of the SGI1 island by mapping the short nucleotide reads of each genome to the SGI1-K reference sequence (NCBI database access number AY463797.8). In the comparison set, we also included the genomes of S. Kentucky, which were characterized by different SGI1-K island variants, as well as other derived SGI1-K island variants, SGI1-P and SGI-Q [5].
Results
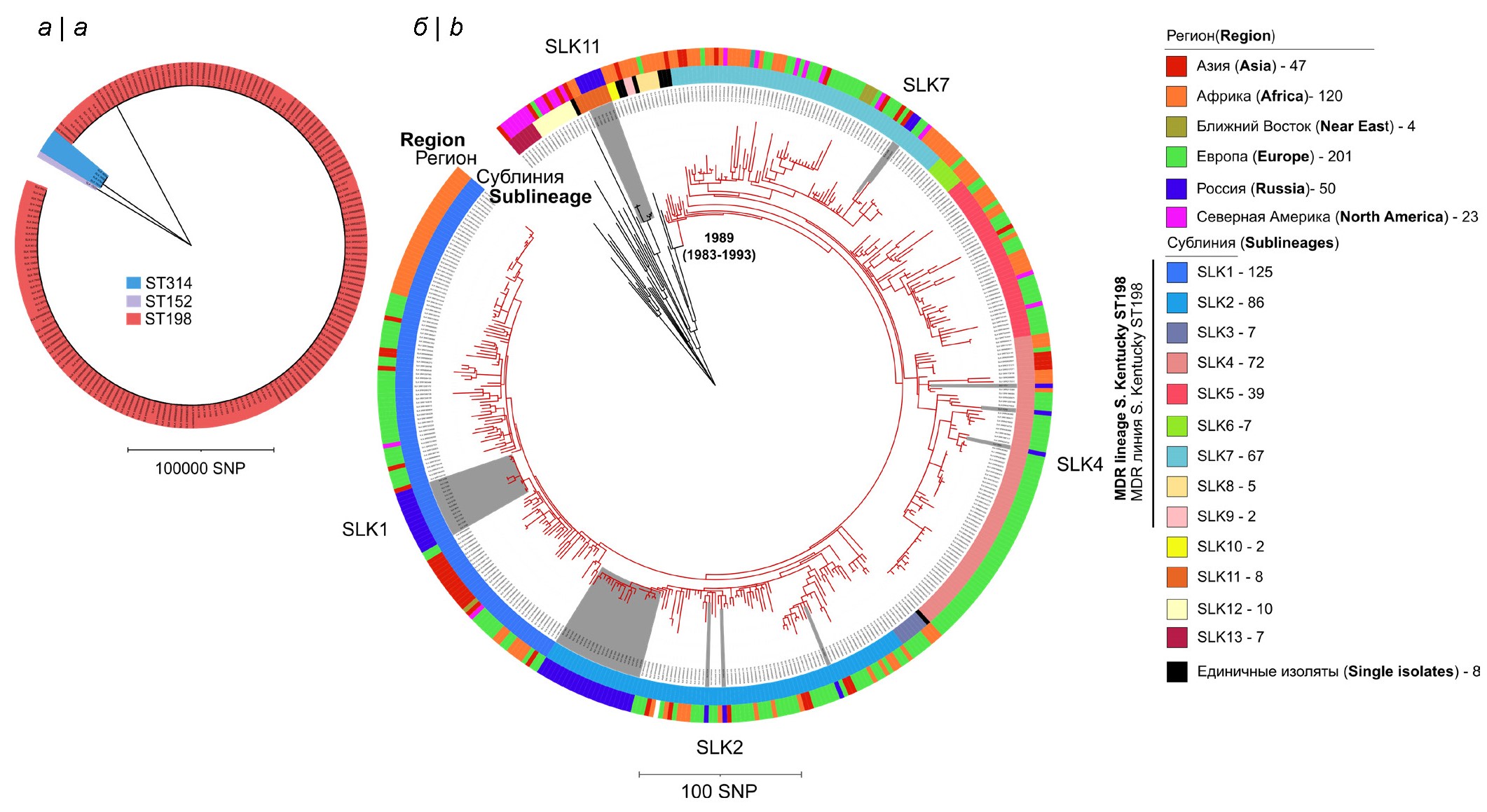
According to the results of the analysis, the majority of strains (n = 50) belonged to ST198 (90.9%), 4 (7.3%) strains were represented by ST314 and only 1 (1.8%) strain was ST152. Strains with these STs belonged to separate phylogenetic lineages with a high level of genetic divergence at the level of core SNP indicating the polyphyletic nature of this serotype, i.e., the ancestral strains of these STs evolutionarily acquired the same O-antigens through convergent evolution (Fig. 1, a) [25]. Since the majority of the Russian strains belonged to the ST198 genotype, further analysis focused on the detailed analysis of this group. For comparison, we used the previously sequenced genomes of S. Kentucky ST198 that are deposited in international databases (n = 390). These strains were characterized by a wide timeframe (1937–2022), geographic range of strain isolation (5 continents), and belonged to diverse sources (human, food, animal and environmental).

Fig. 1. Мaximum likelihood phylogenetic tree.
а — tree reconstructed based on SNPs of 173 S. Kentucky strains of sequence types ST314, ST152, and ST198. Strains belonging to individual sequence types belong to separate phylogenetic lineages with a high level of genetic divergence; b — maximum likelihood phylogenetic tree reconstructed based on SNPs of 445 S. Kentucky sequence-type ST198 strains. The genome of strain 98K (accession number SRR6898532), isolated in the USA in 1937 from chicken, was used as an outgroup. The clade shown in red indicates the MDR lineage S. Kentucky ST198, which includes nine sublineages SLK1-SLK9, identified in the Fastbaps program. The date of MDR lineage origin (1989 year) is indicated according to J. Hawkey et al., 2019 [5]. The outer sectors of different colors represent the geographic regions in which the strains were isolated. Gray sectors on the tree highlight 50 strains of S. Kentucky ST198 isolated in Russia.
Phylogenetic analysis of 440 ST198 strains allowed us to identify 13 sublineages (BAPS genetic clusters) represented by more than 1 strain. At the same time, 9 sublineages belonged to the international monophyletic MDR lineage of S. Kentucky ST198, and 4 occupied a basal position relative to it (Fig. 1, b). The basal clades of each sublineage of the international MDR clone included strains isolated in African countries, supporting the hypothesis of the origin of the MDR lineage S. Kentucky ST198 on the African continent and subsequent spread throughout the world.
Of the 50 Russian ST198 strains, 44 strains belonged to the international monophyletic MDR lineage S. Kentucky ST198 and belonged to 4 separate sublineages (SLK-1, SLK-2, SLK-4, SLK-7), while 6 strains belonged to the SLK-11 sublineage and occupied a basal position in relation to the MDR lineage of S. Kentucky ST198 (Fig. 1, b). The largest number of Russian strains of S. Kentucky (n = 25) belonged to the SLK-2 subline, the smallest – to the SLK-7 sublineage (n = 2). Note that the SLK-4 sublineage was mainly represented by strains from Europe.
Phylogenetic analysis and geographical region of isolation of S. Kentucky strains in Russia did not reveal any correlations. At the same time, Russian strains of the SLK-2 sublineage are characterized by predominant isolation in the North Asian part of Russia (Omsk and Irkutsk regions, Republic of Buryatia, Krasnoyarsk and Altai krais), while for the SLK-11 sublineage – in European Russia (Moscow and Tula regions), and strains belonging to sublineages SLK-1, SLK-7 and SLK-4 were found in both territories.
Analysis of genetic clustering of Russian strains of S. Kentucky at the level of differences within 5 SNPs allowed us to identify 6 t5-SNP clusters that belonged to 3 sublineages (Fig. 2). Two t5-clusters were associated with the SLK-2 sublineage. One of the t5-clusters included 8 strains that were isolated from different sources (human, environmental and food) during 2019–2020. The second cluster included 2 strains isolated from humans in 2022.

Fig. 2. Genetic diversity and population structure of Russian strains of S. Kentucky ST198 (n = 50).
Maximum likelihood phylogenetic tree reconstructed based on SNP alignment of 51 S. Kentucky sequence-type ST198 strains. The genome of the S. Kentucky strain 93-6429 (short read archive accession number SRR6898537), isolated from a human in 1993 in Indonesia, was used as an outgroup. Individual clades on the phylogenetic tree correspond to the identified sublineages of Russian S. Kentucky isolates. The diagram shows isolate information: geographic region of isolation, source of strain, the epidemiological situation (outbreak/sporadic), identified genetic clusters at the level of differences in 5 SNPs (t5-SNP clusters), as well as information about the presence or the absence of genetic determinants of antibiotic resistance and plasmid replicons.
Russian strains of S. Kentucky SLK-1 sublineage strains also included 2 separate t5-SNP clusters, to which 14 strains belonged. The first cluster included 12 strains isolated from the environment of the poultry farm and turkey during the enzootic of salmonellosis in poultry, thus confirming the clonal relationship of salmonella cases detected at the poultry farm. The second cluster included 2 sporadic strains from humans in 2018 and 2019.
SLK-11 sublineage was characterized by 2 t5-SNP clusters: 1 cluster was for strains isolated from turkey feed in 2020 and the other cluster was for strains isolated from turkey litter at the poultry farm in 2022.
Genetic determinants of S. Kentucky ST198 antibiotic resistance and plasmid diversity
All ST198 strains isolated in Russia contained genes or mutations responsible for antibiotic resistance. Moreover, 62% of the strains contained a total of 6–11 resistance genes. A total of 320 genes and mutations responsible for antimicrobial resistance were identified among the 50 strains. The most frequent point mutations were in the QRDR region determining resistance to quinolones, namely in the gyrA (S83F) genes — 13.8% and parC (S80I) — 13.8%. The tetA tetracycline resistance gene was found in 11.9% of strains. The blaTEM-1, aac(3)-Id and aadA7 genes were present in 5% of strains.
When analyzing various combinations of genetic determinants of resistance, 14 combinations were found. The most frequent (26%) was the combination gyrA(S83F, D87Y)-parC(S80I)-blaTEM-1-sul1-aac(3)-Id-aadA7-tet(A), gyrA(S83F, D87N)-parC(S80I)-sul1-tet(A) and gyrA(S83F, D87N)-parC(S80I)-dfrA14-sul1-sul2-aph(6)-Id-aph(3'')-Ib-tet(A) combinations ranked 2nd and 3rd in frequency of occurrence (8% each).
The nature of the set of identified resistance genes and mutations was reasonably consistent with the phylogenetic clustering of the strains studied. All strains of SLK-2 sublineage were characterized by the presence of 3 chromosomal mutations (gyrA (S83F, D87N) and parC(S80I)), and the genes dfrA14, sul1, sul2, aph(6)-Id, aph(3'')-Ib and tetA were found in the majority of strains. At the same time, bla and aadA genes were absent in the group of SLK-2 strains after 2015 in comparison with others.
Russian strains of SLK-1 also carried 2 mutations in the QRDR region, gyrA(S83F) and parC(S80I), but differed from SLK-2 by a mutation in the gyrA(D87Y) gene. All strains carried the same set of resistance genes blaTEM-1-sul1-sul1-aac(3)-Id-aadA7-tet(A).
The 2 strains of SLK-7 sublineage, like SLK-2, were characterized by similar mutations in the gyrA(S83F, D87N) and parC(S80I) genes. At the same time, strain SLK_5298 included the entire set of genes characteristic of the SGI1-K genomic island (blaTEM-1-sul1-aac(3)-Id-aph(6)-Id-aph(3'')-Id-aph(3'')-Ib-aadA7-tet(A)) [19], and strain SLK_10077 was characterized by the absence of resistance genes.
SLK-4 sublineage strains were characterized by set of mutations in gyrA(S83F, D87G) and parC(S80I). Moreover, 2 strains (SLK_10358 and SLK_4955) carried the blaCTX-M-14 gene, in addition to other genes encoding antibiotic resistance.
In comparison with the other strains, only the plasmid-mediated resistance gene qnrB19 was detected in representatives of the SLK-11 sublineage. No mutations in the QRDR-region and other resistance genes were detected.
Analysis of plasmid diversity allowed us to determine the presence of 13 known types of plasmid incompatibility among sequenced Russian isolates. Col156, rep_cluster_2335 and rep_cluster_2350 were the most frequent. The greatest diversity of plasmids was characteristic of SLK-2 sublineage strains, which, in addition to the above-mentioned plasmids, included IncX, Inc-gamma and ColpVC groups.
SGI1 genomic island analysis
The presence of SGI1 island genes in the sequenced genomes was considered as evidence of SGI1 embedding into the bacterial chromosome, namely in the region from the 3'-end of the trmE gene to the 5'-end of the yidY gene. The results of SGI1-K reference sequence overlap analysis for Russian S. Kentucky strains are presented in Fig. 3.

Fig. 3. The degree and nature of overlap of the reference sequence SGI1-K (NCBI accession number AY463797.8) with short nucleotide reads of each of the compared genomes.
The order of strains in the diagram corresponds to the order of strains in the phylogenetic tree in Fig. 2.
The main differences in SGI1-K variants consist in the variation not only in the composition of the island genes, but also in the set of transposons. In particular, the reference SGI1-K carries a set of 5 transposons Int4-Tn21-Tn1721-Tn5393-Tn3, while the previously described SGI1-K variant in strain 08-KS6 is characterized by the absence of transposon Tn5393 and shortened variants of transposon Tn1721 (Int4-Tn21-∆Tn1721-Tn3), strain 08-5707 has a shortened Tn5393 transposon (Int4-Tn21-Tn1721-∆Tn5393-Tn3), and strain 07-1511 has only 3 transposons (Tn1721-Tn21-Tn3) with region inversion.
The SLK-11 sublineage strains were characterized by the absence of both SGI1 island genes and mobile genetic elements Int4, Tn21, Tn1721, and Tn3, which are characteristic of SGI1-K, indicating the absence of SGI1 island integration into the chromosome.
For strains of SLK-2 sublineage, the SGI1-K reference sequence overlap ranged from 19% to 61%. At the same time, most strains were characterized by the absence of the main part of the backbone genes in the range from the S005 gene to the resG gene and the presence of transposons Int4, Tn21, Tn1721 carrying resistance genes. SLK_7836, SLK_7843 and SLK_7842 strains, which lacked mobile elements, were also characterized by the absence of antibiotic resistance genes. SLK_6643 strain was characterized by a similar pattern of reference sequence overlap with SGI1-K. In turn, SLK_5116 and SLK_4223 strains were similar to the SGI1-K 08-5707 variant with a deletion of the bark traG-resG genes and a set of Int4-Tn21-Tn1721-Tn3 mobile elements.
All SLK-1 sublineage strains had a similar composition of SGI1 island genes and 90% overlap of the SGI1-K reference sequence. The set of mobile elements, including Int4-Tn21-Tn1721-Tn3, was similar to the SGI1-K variant described previously in strain 08-KS6.
In the SLK-7 sublineage, strain SLK_10077 was characterized by 60% percent overlap of the reference sequence due to the presence of island genes and absence of mobile elements, which correlated with the absence of resistance genes in the contig analysis. At the same time, the nucleotide reads of strain SLK_5298 overlapped the reference sequence SGI1-K by 100%.
SLK-4 sublineage strains were characterized by different variants of SGI1. For example, strain SLK_10358 (61% overlap) was characterized by the presence of all island genes, while only transposon Tn5393 was present among mobile elements. SLK_4955 (87% overlap) and SLK_1731 (85% overlap) strains were characterized by a similar set of Int4-Tn21-Tn1721-Tn539 transposons. However, SLK_1731 lacked the SGI1 island genes (S025-resG).
Discussion
The epidemiologic significance of S. Kentucky serotype is due to its potential for spread, adaptability to various environmental conditions, and genetic mechanisms, including chromosomal and plasmid-mediated, which allow resistance to a wide range of antimicrobial drugs to be realized. Along with a high proportion of strains resistant to ciprofloxacin, cases of combined resistance to ciprofloxacin and cefotaxime (III generation cephalosporins) have been registered. In particular, strains with combined resistance were registered on the island of Malta and belonged to bacterial isolates producing ESBL [26]. S. Kentucky strains with genes for ESBL (CTX-M-1, -M-14, -M-15, -M-104), cephalosporinase AmpC beta-lactamases (CMY-2 and -4), and carbapenemases (VIM-2, OXA-48, NDM-1) localized not only on plasmids but also in the SGI1 chromosomal region [4, 5, 19, 26–29]. Although salmonellosis is considered a strictly zoonotic infection, there have been suggestions of the spread of some epidemic clones of S. Kentucky from human to human and that humans are a reservoir [18].
To date, there are 10 different STs of S. Kentucky, 3 of which (ST198, ST152, and ST314) are the most frequent [30]. In certain cases, different representation of these STs has been noted, both territorially (international differences) and in relation to the sources of Salmonella isolation [31, 32]. However, the predominance of ST198 and ST152 in humans and poultry has been noted [33]. In our study, more than 90% of the strains associated with different isolation sources were ST198 and ST152 was represented by a single strain, which is consistent with international data.
The use of whole genome sequencing is the gold standard and universal tool for genomic surveillance of socially important pathogens [34, 35]. The possibility of hierarchical clustering of phylogenetically significant markers of a certain pathogen at different levels of detail and comparison with sequences deposited in international databases not only provides valuable information on evolutionary trends and assessment of pathogen spreading factors on a global scale, but also makes it possible to determine clonal and familial relationships between strains for epidemiological investigation of outbreak and sporadic morbidity [36–38].
According to the phylogenetic analysis, the majority of ST198 strains circulating in Russia belonged to 4 major sublineages of the international monophyletic MDR lineage of S. Kentucky ST198, which includes strains with MDR phenotype to antimicrobials [5]. A majority of Russian strains belonged to phylogenetic sublineages SLK-1 and SLK-2. At the same time, the long period of isolation of strains from different sources belonging to these two sublineages may indicate the presence of two established and genetically distinct subpopulations of the MDR-lineage of S. Kentucky ST198 on the territory of Russia. It is interesting to note that the character of phylogenetic clustering into separate sublineages coincided with a certain combination of 3 mutations in the gyrA and parC genes, determining resistance to quinolones, which allows us to use them as markers for differentiation of Russian S. Kentucky ST198 strains into the main phylogenetic sublineages.
Two strains of the SLK-4 sublineage, SLK_10358 (Irkutsk, sporadic case in 2022) and SLK_4955 (a representative strain from a salmonellosis outbreak in Izhevsk in 2015), were representatives of the phylogenetic sublineage that has been associated with the S. Kentucky population established since 2005 in European countries [18]. The peculiarity of these ciprofloxacin-resistant S. Kentucky strains is the presence of the blaCTX-M-14 gene integrated into the chromosome, encoding ESBL [18]. This gene was also detected in Russian strains of SLK-4 sublineage. Cases of isolation of such strains may indicate single and independent cases of introduction of strains of S. Kentucky blaCTX-M-14 from the territory of the EU countries, but this assumption requires investigation of an expanded sample of strains.
The data obtained, based on the identification of genetically similar strains, using a clustering approach at a level not exceeding 5 nucleotide variations between strains (t5-SNP cluster), revealed 6 groups of clonally related strains within 3 sublineages of Russian S. Kentucky ST198 strains. The size of the groups varied from 2 to 11 strains. In most cases, the identified t5 clusters were associated with the circulation of a particular clone of S. Kentucky ST198 in a limited area and in a relatively short period of time (not more than 1 year): in an epizootic outbreak of salmonellosis at a turkey poultry farm in Tula region (2012); detection of salmonella strains in turkey litter at a poultry farm in Moscow region (2022); in mixed feed for turkeys in the Moscow region (2022); and in 2 independent cases of salmonellosis in humans in Angarsk and Irkutsk in 2022. At the same time, our studies demonstrate the possibility of the existence of latent circulation of a certain clone of S. Kentucky ST198 with strains detected in 2019 and 2020 in Omsk (n = 7) and Irkutsk (n = 1), which were associated with sporadic cases of salmonellosis and isolation of salmonellae from environmental objects and from food products, thus indicating a probable epidemiologic linkage.
The approach used in our study to analyze the structure of the SGI1 genomic island allowed us to indirectly assess its composition and demonstrate the sensitivity of this genomic region to genetic rearrangements caused by the activity of transpositional elements [5, 9, 19]. These rearrangements can lead to the deletion of some or all genes within SGI1 [5]. In most cases, Russian strains are characterized by the presence of SGI1-K variants. Nevertheless, in a number of genomes, the detected set of transposons differed from the existing SGI1 variants, which probably indicates the presence of a new variant of the genomic island. High variation of this island was also observed in previous studies, where almost every strain was characterized by a different SGI1 structure. In addition to large deletions of the SGI1 island, some strains had inversions of all or part of the segment including resistance genes, as well as transposon rearrangements [5].
The analysis of SGI1 organization and phylogenetic clustering of strains based on the analysis of SNP profiles generally correlated with each other for different SLK sublineages. However, in SLK-2 strains belonging to the same t5-SNP cluster, the SGI1 structure showed differences in set of transposons. Such discrepancies may indicate a high rate of genetic rearrangements of the SGI1 region in clonally related strains and explain the difference in antibiotic resistance gene set.
The presence of SGI1 correlated with the detected antibiotic resistance gene set. Most strains (86%) were characterized by the presence of aadA7, blaTEM-1, sul1 and tetA genes, which are known to be associated with SGI1 in S. Kentucky ST198 strains [19].
Collectively, the antimicrobial resistance genes identified were responsible for resistance to various classes of antibiotics, including aminoglycosides, β-lactams, phenicols, quinolones, sulfonamides and tetracyclines. Based on the data obtained, there is still uncertainty about the association of certain resistance genes with the detected plasmid type due to the lack of completed genome assembly. However, the data obtained on the diversity of plasmid types indicate their ability to transfer resistance genes. It has been shown that the small ColRNAI plasmids, rep_cluster_2335 and rep_cluster_2350 can successfully carry a diverse range of resistance genes, such as blaCTX-M-15, blaSHV-11, blaTEM-1B, blaOXA-1, ac(3)-IIa, strB, strA, aadA16, qnrB66, oqxA and oqxB [39–42].
Conclusion
Despite the relatively small sample of strains, we were able not only to come closer to understanding the population structure of Russian S. Kentucky ST198 strains on a global scale, but also to conduct a detailed study of the genetic determinants of antibiotic resistance, including the structure of the SGI1 genomic island. The findings provide a basis for understanding and tracking the ongoing evolution of the MDR lineage, which is a globally distributed clone capable of rapid expansion and accumulation of antimicrobial resistance determinants. Our data demonstrate instances of circulating clonally related S. Kentucky ST198 strains in different sources, which indicates the need to develop an integrated approach to salmonellosis surveillance based on the One Health concept.
1 Information bulletins of the reference Center for Monitoring of Salmonellosis No. 35–26. URL: https://www.epid-oki.ru/otchety.html
2 BBTools. URL: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/ (дата обращения 12.12.2023)
3 EnteroBase. Quality Assessment evaluation. URL: https://enterobase.readthedocs.io/en/latest/pipelines/backend-pipeline-qaevaluation.html (дата обращения 12.12.2023).
4 mlst: scan contig files against traditional PubMLST typing schemes. URL: https://github.com/tseemann/mlst (дата обращения 12.12.2023).
About the authors
Konstantin V. Kuleshov
Central Research Institute for Epidemiology
Author for correspondence.
Email: konstantinkul@gmail.com
ORCID iD: 0000-0002-5238-7900
Cand. Sci. (Biol.), Head, Laboratory of molecular diagnostics and epidemiology of enteric infections
Russian Federation, MoscowAnastasia S. Pavlova
Central Research Institute for Epidemiology
Email: a.pavlova@cmd.su
ORCID iD: 0000-0003-4619-9337
researcher, Laboratory of molecular diagnostics and epidemiology of enteric infections
Russian Federation, MoscowAnna A. Kremleva
Federal Center for Animal Health
Email: viktoriya1409@yandex.ru
ORCID iD: 0000-0002-6290-6639
researcher, Department of bacteriology of the Testing central scientific and methodological veterinary laboratory
Russian Federation, MoscowAnna E. Karpenko
Central Research Institute for Epidemiology
Email: a.egorova@cmd.su
ORCID iD: 0000-0003-0486-1353
researcher, Laboratory of molecular mechanisms of antibiotic resistance
Russian Federation, MoscowYuliya V. Mikhaylova
Central Research Institute for Epidemiology
Email: mihailova@cmd.su
ORCID iD: 0000-0002-5646-538X
Cand. Sci. (Biol.), Head, Laboratory of molecular mechanisms of antibiotic resistance
Russian Federation, MoscowNatalia E. Krutova
Central Research Institute for Epidemiology
Email: nkrutova@cmd.su
ORCID iD: 0000-0003-2925-5376
junior researcher, Laboratory of molecular diagnostics and epidemiology of enteric infections
Russian Federation, MoscowMaria R. Lisitsyna
Central Research Institute for Epidemiology
Email: kopylova@cmd.su
ORCID iD: 0009-0009-2344-6383
junior researcher, Laboratory of molecular diagnostics and epidemiology of enteric infections
Russian Federation, MoscowKristina R. Popova
Central Research Institute for Epidemiology
Email: c.bicmetowa@yandex.ru
ORCID iD: 0000-0003-3368-7833
junior researcher, Laboratory of molecular diagnostics and epidemiology of enteric infections
Russian Federation, MoscowOlga A. Veselova
Central Research Institute for Epidemiology
Email: sapienscri@gmail.com
ORCID iD: 0000-0002-5041-4370
researcher, Laboratory of molecular diagnostics and epidemiology of enteric infections
Russian Federation, MoscowAlexandr T. Podkolzin
Central Research Institute for Epidemiology
Email: apodkolzin@pcr.ru
ORCID iD: 0000-0002-0044-3341
D. Sci. (Med.), Deputy Director for epidemiology
Russian Federation, MoscowVasily G. Akimkin
Central Research Institute for Epidemiology
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
D. Sci. (Med.), Professor, Academician of the Russian Academy of Sciences, Director
Russian Federation, MoscowReferences
- Koutsoumanis K., Allende A., Alvarez-Ordóñez A., et al. Salmonella control in poultry flocks and its public health impact. EFSA J. 2019;17(2):e05596. DOI: https://doi.org/10.2903/j.efsa.2019.5596
- Le Hello S., Harrois D., Bouchrif B., et al. Highly drug-resistant Salmonella enterica serotype Kentucky ST198-X1: a microbiological study. Lancet Infect. Dis. 2013;13(8):672–9. DOI: https://doi.org/10.1016/S1473-3099(13)70124-5
- Le Hello S., Weill F.X., Guibert V., et al. Early strains of multidrug-resistant Salmonella enterica serovar Kentucky sequence type 198 from Southeast Asia harbor Salmonella genomic island 1-J variants with a novel insertion sequence. Antimicrob. Agents Chemother. 2012;56(10):5096–102. DOI: https://doi.org/10.1128/AAC.00732-12
- Le Hello S., Hendriksen R.S., Doublet B., et al. International spread of an epidemic population of Salmonella enterica serotype Kentucky ST198 resistant to ciprofloxacin. J. Infect. Dis. 2011;204(5):675–84. DOI: https://doi.org/10.1093/infdis/jir409
- Hawkey J., Le Hello S., Doublet B., et al. Global phylogenomics of multidrug-resistant Salmonella enterica serotype Kentucky ST198. Microb. Genom. 2019;5(7):e000269. DOI: https://doi.org/10.1099/mgen.0.000269
- Bie L., Fang M., Li Z., et al. Identification and characterization of new resistance-conferring SGI1s (Salmonella genomic island 1) in Proteus mirabilis. Front. Microbiol. 2018;9:3172. DOI: https://doi.org/10.3389/fmicb.2018.03172
- Doublet B., Golding G.R., Mulvey M.R., Cloeckaert A. Secondary chromosomal attachment site and tandem integration of the mobilizable Salmonella genomic island 1. PLoS One. 2008; 3(4):e2060. DOI: https://doi.org/10.1371/journal.pone.0002060
- Le Hello S. Bekhit A., Granier S.A., et al. The global establishment of a highly-fluoroquinolone resistant Salmonella enterica serotype Kentucky ST198 strain. Front. Microbiol. 2013;4:395. DOI: https://doi.org/10.3389/fmicb.2013.00395
- Cohen E., Davidovich M., Rokney A., et al. Emergence of new variants of antibiotic resistance genomic islands among multidrug-resistant Salmonella enterica in poultry. Environ. Microbiol. 2020;22(1):413–32. DOI: https://doi.org/10.1111/1462-2920.14858
- Beutlich J., Guerra B., Schroeter A., et al. Highly ciprofloxacin resistant Salmonella enterica serovar Kentucky isolates in turkey meat and a human patient. Berl. Munch. Tierarztl. Wochenschr. 2012;125(3-4):89–95. (in German)
- Münch S., Braun P., Wernery U., et al. Prevalence, serovars, phage types, and antibiotic susceptibilities of Salmonella strains isolated from animals in the United Arab Emirates from 1996 to 2009. Trop. Anim. Health Prod. 2012;44(7):1725–38. DOI: https://doi.org/10.1007/s11250-012-0130-4
- Wasyl D., Hoszowski A. First isolation of ESBL-producing Salmonella and emergence of multiresistant Salmonella Kentucky in turkey in Poland. Food Res. Int. 2012;45(2):958–61. DOI: https://doi.org/10.1016/j.foodres.2011.07.024
- Павлова А.С., Кулешов К.В., Крутова Н.Е. и др. Характеристика антибиотикорезистентности нетифоидных сальмонелл, циркулирующих на территории Российской Федерации в период с 2019 по 2022 год. Журнал микробиологии, эпидемиологии и иммунобиологии. 2023;100(5):287–301. Pavlova A.S., Kuleshov K.V., Krutova N.E., et al. Characteristics of antibiotic resistance of non-typhoidal Salmonella circulating in the Russian Federation in the period from 2019 to 2022. Journal of Microbiology, Epidemiology and Immunobiology. 2023;100(5):287–301. DOI: https://doi.org/ https://doi.org/10.36233/0372-9311-451 EDN: https://elibrary.ru/tmxvam
- Кафтырева Л.А., Егорова С.А., Макарова М.А. Детекция международных клонов высокого риска Salmonella и Escherichia coli – возбудителей заболеваний, передающихся с пищевыми продуктами, в Российской Федерации. Инфекция и иммунитет. 2020;10(3):565–9. Kaftyreva L.A., Egorova S.A., Makarova M.A. Detection of international high-risk clones of food-borne pathogens Salmonella and Escherichia coli in the Russian Federation. Russian Journal of Infection and Immunity. 2020;10(3):565–9. DOI: https://doi.org/ https://doi.org/10.15789/2220-7619-DOI-150 EDN: https://elibrary.ru/iybhmg
- Souvorov A., Agarwala R., Lipman D.J. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018;19(1): 153. DOI: https://doi.org/10.1186/s13059-018-1540-z
- Yoshida C.E., Kruczkiewicz P., Laing C.R., et al. The Salmonella In Silico Typing Resource (SISTR): an open web-accessible tool for rapidly typing and subtyping draft salmonella genome assemblies. PLoS One. 2016;11(1):e0147101. DOI: https://doi.org/10.1371/journal.pone.0147101
- Zhang S., den Bakker H.C., Li S., et al. SeqSero2: rapid and improved Salmonella serotype determination using whole-genome sequencing data. Appl. Environ. Microbiol. 2019;85(23):e01746-19. DOI: https://doi.org/10.1128/AEM.01746-19
- Coipan C.E., Westrell T., van Hoek A.H.A.M., et al. Genomic epidemiology of emerging ESBL-producing Salmonella Kentucky bla CTX-M-14b in Europe. Emerg. Microbes Infect. 2020;9(1):2124–35. DOI: https://doi.org/10.1080/22221751.2020.1821582
- Mashe T., Thilliez G., Chaibva B.V., et al. Highly drug resistant clone of Salmonella Kentucky ST198 in clinical infections and poultry in Zimbabwe. npj Antimicrob. Resist. 2023;1(1):6. DOI: https://doi.org/10.1038/s44259-023-00003-6
- Dallman T., Ashton P., Schafer U., et al. SnapperDB: a database solution for routine sequencing analysis of bacterial isolates. Bioinformatics. 2018;34(17):3028–9. DOI: https://doi.org/10.1093/bioinformatics/bty212
- Croucher N.J., Page A.J., Connor T.R., et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43(3):e15. DOI: https://doi.org/10.1093/nar/gku1196
- Tonkin-Hill G., Lees J.A., Bentley S.D., et al. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019;47(11):5539–49. DOI: https://doi.org/10.1093/nar/gkz361
- Feldgarden M., Brover V., Gonzalez-Escalona N., et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021;11(1):12728. DOI: https://doi.org/10.1038/s41598-021-91456-0
- Robertson J., Nash J.H.E. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018;4(8):e000206. DOI: https://doi.org/10.1099/mgen.0.000206
- Timme R.E., Pettengill J.B., Allard M.W., et al. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol. Evol. 2013;5(11):2109–23. DOI: https://doi.org/10.1093/gbe/evt159
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017. EFSA J. 2019;17(2):e05598. DOI: https://doi.org/10.2903/j.efsa.2019.5598
- Lei C.W., Zhang Y., Wang X.C., et al. Draft genome sequence of a multidrug-resistant Salmonella enterica serotype Kentucky ST198 with chromosomal integration of blaCTX-M-14b isolated from a poultry slaughterhouse in China. J. Glob. Antimicrob. Resist. 2020;20:145–6. DOI: https://doi.org/10.1016/j.jgar.2019.12.006
- Chen H., Song J., Zeng X., et al. National prevalence of Salmonella enterica serotype Kentucky ST198 with high-level resistance to ciprofloxacin and extended-spectrum cephalosporins in China, 2013 to 2017. mSystems. 2021;6(1):e00935-20. DOI: https://doi.org/10.1128/mSystems.00935-20
- El Hage R., Losasso C., Longo A., et al. Whole-genome characterisation of TEM-1 and CMY-2 β-lactamase-producing Salmonella Kentucky ST198 in Lebanese broiler chain. J. Glob. Antimicrob. Resist. 2020;23:408–16. DOI: https://doi.org/10.1016/j.jgar.2020.11.002
- Achtman M., Wain J., Weill F.X., et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012;8(6):e1002776. DOI: https://doi.org/10.1371/journal.ppat.1002776
- Slowey R., Kim S.W., Prendergast D., et al. Genomic diversity and resistome profiles of Salmonella enterica subsp. enterica serovar Kentucky isolated from food and animal sources in Ireland. Zoonoses Public Health. 2022;69(1):1–12. DOI: https://doi.org/10.1111/zph.12884
- Wang S., Liao X., Xiong Z., et al. Characterization of the emerging multidrug-resistant Salmonella enterica serotype Kentucky ST314 in China. Zoonoses Public Health. 2021;68(6):622–9. DOI: https://doi.org/10.1111/zph.12850
- Soltys R.C., Sakomoto C.K., Oltean H.N., et al. High-resolution comparative genomics of Salmonella Kentucky aids source tracing and detection of ST198 and ST152 lineage-specific mutations. Front. Sustain. Food Syst. 2021;5:695368. DOI: https://doi.org/10.3389/fsufs.2021.695368
- Deng X., den Bakker H.C., Hendriksen R.S. Genomic epidemiology: whole-genome-sequencing-powered surveillance and outbreak investigation of foodborne bacterial pathogens. Annu. Rev. Food Sci. Technol. 2016;7:353–74. DOI: https://doi.org/10.1146/annurev-food-041715-033259
- Franz E., Gras L.M., Dallman T. Significance of whole genome sequencing for surveillance, source attribution and microbial risk assessment of foodborne pathogens. Curr. Opin. Food Sci. 2016;8:74–9. DOI: https://doi.org/10.1016/j.cofs.2016.04.004
- Li S., He Y., Mann D.A., Deng X. Global spread of Salmonella Enteritidis via centralized sourcing and international trade of poultry breeding stocks. Nat. Commun. 2021;12(1):5109. DOI: https://doi.org/10.1038/s41467-021-25319-7
- Pijnacker R., Dallman T.J., Tijsma A.S.L., et al. An international outbreak of Salmonella enterica serotype Enteritidis linked to eggs from Poland: a microbiological and epidemiological study. Lancet Infect. Dis. 2019;19(7):778–86. DOI: https://doi.org/10.1016/S1473-3099(19)30047-7
- Chattaway M.A., Dallman T.J., Larkin L., et al. The transformation of reference microbiology methods and surveillance for Salmonella with the use of whole genome sequencing in England and Wales. Front. Public Health. 2019;7:317. DOI: https://doi.org/10.3389/fpubh.2019.00317
- Agyepong N., Govinden U., Owusu-Ofori A., et al. Genomic characterization of multidrug-resistant ESBL-producing Klebsiella pneumoniae isolated from a Ghanaian teaching hospital. Int. J. Infect. Dis. 2019;85:117–23. DOI: https://doi.org/10.1016/j.ijid.2019.05.025
- Lerminiaux N., Mitchell R., Bartoszko J., et al. Plasmid genomic epidemiology of blaKPC carbapenemase-producing Enterobacterales in Canada, 2010–2021. Antimicrob. Agents Chemother. 2023;67(12):e0086023. DOI: https://doi.org/10.1128/aac.00860-23
- Nakamura K., Seto K., Lee K., et al. Global population structure, genomic diversity and carbohydrate fermentation characteristics of clonal complex 119 (CC119), an understudied Shiga toxin-producing E. coli (STEC) lineage including O165:H25 and O172:H25. Microb. Genom. 2023;9(3):mgen000959. DOI: https://doi.org/10.1099/mgen.0.000959
- Rehman M.A., Rempel H., Carrillo C.D., et al. Virulence genotype and phenotype of multiple antimicrobial-resistant Escherichia coli isolates from broilers assessed from a «One-Health» perspective. J. Food Prot. 2022;85(2):336–54. DOI: https://doi.org/10.4315/JFP-21-273
Supplementary files

