


Vol 99, No 4 (2022)

- Year: 2022
- Published: 27.09.2022
- Articles: 13
- URL: https://microbiol.crie.ru/jour/issue/view/51
Full Issue

ORIGINAL RESEARCHES
COVID-19: evolution of the pandemic in Russia. Report II: dynamics of the circulation of SARS-CoV-2 genetic variants

Abstract
Background. The ongoing pandemic of the novel coronavirus infection (COVID-19) draws attention to the significance of molecular and genetic monitoring of the SARS-CoV-2 spread among the population of the Russian Federation. The aim of the study was to analyze the dynamics of circulation of SARS-CoV-2 genetic variants in Russia.
Materials and methods. The analysis of the circulation dynamics for SARS-CoV-2 genetic variants in Russia was carried out, covering the period from 28/12/2020 to 26/6/2022. The analysis included the data from Rospotrebnadzor Report No. 970 "Information about Infectious Diseases in Individuals with Suspected Novel Coronavirus Infection" and the Virus Genome Aggregator of Russia (VGARus). The presence of SARS-CoV-2 RNA was confirmed by the real-time reverse transcription polymerase chain reaction. The primer panels developed at the Central Research Institute of Epidemiology were used for amplification of genomic fragments and the subsequent sequencing.
Results and discussion. Using the Russian VGARus platform developed by the Central Research Institute of Epidemiology, we received the data on mutational variability of SARS-CoV-2. By monitoring the circulation of SARS-CoV-2 genetic variants in Russia from 28/12/2020 to 26/6/2022, we found that Delta and Omicron genetic variants prevailed at different stages of the epidemic.
Conclusion. The data of molecular and genetic studies are an essential component of epidemiological surveillance, being critically important for making executive decisions aimed at prevention of further spread of SARS-CoV-2 and laying the groundwork for creating new vaccines.
 381-396
381-396



Features of the spread of a new coronavirus infection in the territory of municipalities of the Rostov region
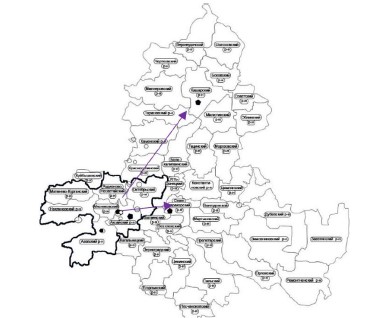
Abstract
Introduction. Since the registration of the first cases of COVID-19 in the PRC, due to the high migration activity of the population, the new coronavirus infection has spread throughout the world, including the Russian Federation.
Aim. To establish the features of the spread of a new coronavirus infection in the Rostov Region. Materials and methods.The analysis of 81 cases of the importation of a new coronavirus infection by persons who arrived in the Rostov Region from abroad or other regions of the Russian Federation was carried out based on the data of the Rostov Region office of the Federal Service for Supervision of Consumer Protection and Human Welfare. The dynamics of the spread of COVID-19 in the administrative territories of the Rostov Region has been analyzed. The data of viral genome-wide sequencing (n = 155) carried out in the Rostov-on-Don Research Anti-Plague Institute were used in this study.
Results. In the period from 03/21/2020 to 03/28/2020, cases imported both from abroad and from other regions of the Russian Federation were registered in the Rostov Region, , mainly on the territory of the Rostov urban agglomeration. The vector of the spread of the disease was directed from the administrative center of the region to the periphery. The emergence of a new genetic line B.617.2 (Delta) probably led to a significant increase in the incidence in the Rostov Region.
Conclusions. The spread of a new coronavirus infection in the Rostov Region was facilitated by the one of the main social factors of epidemiological risk, the population migration, which led to the importation of the infection to the administrative center of the subject, Rostov-on-Don city. Taking into account peculiarities of the Rostov region, the largest proportion of COVID-19 cases was recorded in the Rostov urban agglomeration. Against the background of the dominance of the “Delta” variant of the virus on the territory of the Rostov region, there was a tendency towards an increase in the number of cases.
410-419
Biological characterization of cold-adapted SARS-CoV-2 variants
Abstract
Introduction. The emergence of new epidemiologically significant variants of SARS-CoV-2 has shifted emphasis to development of a live vaccine, which would be able to provide protection against a wide range of antigenic variants of the virus.
The aim of the study was to obtain SARS-CoV-2 variants attenuated through cold adaptation and to provide their biological characterization.
Materials and methods. The Dubrovka laboratory strain of SARS-CoV-2 and its variants were cultured on Vero and Calu-3 cells. The virus quantification was performed by virus titration in Vero cells and by real-time reverse transcription-polymerase chain reaction. SARS-CoV-2 virions were analyzed using transmission electron microscopy. Genome sequences of the virus were identified by nanopore sequencing. The attenuation (att) phenotype of SARS-CoV-2 variants was identified using Syrian hamsters as an animal model for COVID-19.
Results. Cold-adapted (ca) SARS-CoV-2 variants – Dubrovka-ca-B4 and Dubrovka-ca-D2 were produced by continued passaging of the Dubrovka strain in the Vero cell culture at the temperature being gradually decreased to 23ºC and by subsequent cloning. Up to 20 nucleotide substitutions and 18 amino acid substitutions were detected in genomes of ca-variants. Ca-variants, as distinct from the parent Dubrovka strain, actively replicated at 23ºC, while the Dubrovka-ca-D2 variant had a temperature-sensitive (ts) phenotype (did not replicate at 39ºC). Ca-variants of the virus replicated poorly at 37ºC in the Calu-3 human lung cell culture, which, along with the ts-phenotype, can be a marker of virus attenuation for humans. In the intranasally infected Syrian hamsters, ca-variants of the virus demonstrated an attenuation phenotype: they did not cause loss of appetite, fatigue, drowsiness, did not slow down weight gain, replicating much more slowly in the lungs and brain compared to the virulent Dubrovka strain.
Conclusion. The obtained attenuated SARS-CoV-2 ca-variants, Dubrovka-ca-B4 and Dubrovka-ca-D2, should be studied further as candidate vaccine strains for a live attenuated vaccine against COVID-19.
397-409
An in vitro study of interactions of Candida albicans with Klebsiella pneumoniae and Enterococcus faecalis isolated from intestinal microbiome of HIV infected patients
Abstract
The aim: In vitro identification of targets for antagonism factors in klebsiellas and enterococci for Candida albicans isolated from the intestinal microbiome of HIV infected patients.
Materials and methods. The tests were performed using 38 Candida albicans strains, 28 Klebsiella pneumoniae strains, and 30 Enterococcus faecalis strains isolated from the intestinal microbiome of 89 HIV infected children. The mean age of the patients was 24 ± 2 months; the group consisted of 49 (55%) boys and 40 (45%) girls. Microorganisms were isolated from the intestinal biotope using such selective media as HiChrome Candida Agar, HiChrome Klebsiella Selective Agar Base, and Enterococcus Agar; the study included identification of species. Model experiments were performed to study anti-catalase activity of E. faecalis exometabolites and the impact of K. pneumoniae on morphological transformation of C. albicans fungi.
Results. Klebsiellas decrease the intensity of germ tube formation in C. albicans by 58.7% (p < 0.01). When cocultured, 12.3% of the yeast cells produce germ tubes, while 29.8% of transformed cells was detected in the fungal monoculture. It has been found that exometabolites of 65.7% of E. faecalis strains decrease production of catalase in C. albicans. The initial catalase level in untreated cultures of C. albicans averages 1.02 µmol/min of optical density; after they are treated with E. faecalis exometabolites, the level decreases to 0.55 µmol/min, i.e. by 46.1% (p < 0.05).
Conclusions. K. pneumoniae and E. faecalis demonstrate antagonism of different intensity toward C. albicans. Morphological transformation and catalase production are targets for antagonism factors of facultative microbiota in C. albicans.
420-427
On the assesment of the etiological significance of bacteria detected in the male genital tract
Abstract
Introduction. Currently, there is an increasing importance of microbial associations in the pathogenesis of genital inflammatory diseases. However, the issues of deciphering the taxonomic affiliation and the diagnostic significance of the bacteria detected in this case remain unresolved.
The aim of the study was to review the diagnostic significance of the quantitative approach in determining the etiological role of microorganisms in andrology.
Materials and methods. For the study, samples of ejaculate and/or discharge from the urethra from 15 men who were in infertile marriages, 12 with a confirmed diagnosis of "acute genital gonococcal infection" were used. The testing included a classic bacteriological study and the metagenomic analysis of 16S ribosomal RNA samples carried out at the Department of Collection Cultures of the State Research Center for Applied Microbiology (Obolensk). To standardize the distribution of samples into groups based on the indicators of alpha diversity and the concentration of putrescine, the range of variation and the average linear deviation were used. Statistical analysis of the obtained data was carried out using Excel software (MS Office package).
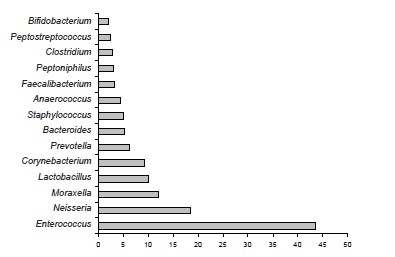
Results. The microbial landscape of the ejaculate was characterized by the predominance of representatives of the genera Enterococcus, Neisseria, Lactobacillus, Corynebacterium, Prevotella, Bacteroides. Stable associations of E. faecalis and M. osloensis were detected in the ejaculate. When using the cultural method, representatives of the genus Moraxella were not isolated in any sample. It was shown that discrepancies can affect not only quantitative indicators, but also reveal inconsistencies between the qualitative assessment of detected genetic markers and the results of identification in a bacteriological study of individual representatives of similar or phenotypically similar taxa.
Discussion. The results of this study indicate that the lesser diversity of opportunistic pathogens gives them more opportunities to realize their pathogenic potential. On the other hand, in a complex community with greater alpha diversity, its realization is hindered by a complex intermicrobial relationships and the need to survive.
Conclusion. As a result, it seems that the most promising approach is the integrated use of cultural methods and metagenomics with a comparative statistical analysis of the obtained qualitative and quantitative indicators.
428-435
Multiplex PCR screening for virulence genes of Klebsiella pneumoniae isolated from microbiota of diseased and healthy people
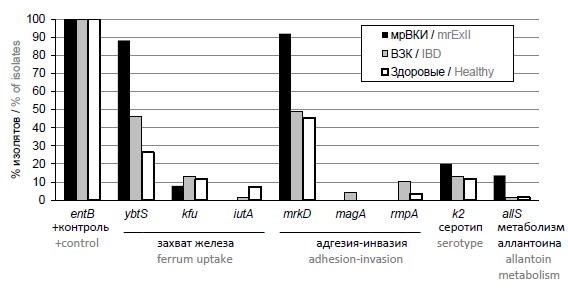
Abstract
Introduction. Klebsiella pneumoniae in human microbiota may appear as a part of commensal microbiota, and as hypervirulent pathogen, for example, hypermucoid pathotype. This pathotype is characterized by certain genetic determinants, leading to the possibility of detecting the pathogenic potential of isolates by PCR.
Aim of the study: to evaluate and compare pathogenic potential of K. pneumoniae isolates from practically healthy people, patients with inflammatory bowel disease (IBD) and extraintestinal infections (ExII).
Materials and methods. Testing was performed with the set of nucleotides for multiplex PCR analysis targeting eight potentially virulent genes with the following functions: ferrum uptake (ybsT, kfu, iutA), adhesion and invasion (mrkD), hypermucoid phenotype and virulent serotypes (mrkD, magA, rmpA, k2) and metabolism of allantoin (allS). PCR assay was used to screen Klebsiella pneumoniae isolates from feces of patients with IBD (69 isolates) and of practically healthy people (68 isolates), and multiresistant isolates from biological material (blood, urine, surgical wounds, bronchoalveolar lavage) of patients with extraintestinal infections (mrExII, 25 isolates).
Results. Results of the testing demonstrated association of four of targeted determinants with the patients diagnoses. YbtS gene was significantly more often found in isolates from IBD (р = 0.024) and mrExII (p < 0.001) groups. RmpA gene was significantly more often detected in IBD group (р = 0.038). Extraintestinal infectious isolates were significantly (р ≤ 0.001) enriched with mrkD and allS genes (р = 0.032).
Conclusion. The most potentially virulent group was isolated from patients with extraintestinal infections, the least virulent — isolates from feces of practically healthy people. The most frequently detected virulence genes were involved in adhesion and hypermucoid phenotype formation.
436-444
Study of microbial factors in exacerbation of chronic rhinosinusitis with nasal polyps
Abstract
Introduction. Chronic rhinosinusitis with nasal polyps (CRSwNP) is considered a multifactorial disease. There are data on the contribution of fungi and viruses to the initiation and development of the inflammatory process, data on the effect of superantigens, biofilms and microbiota on the growth of polyps in the paranasal sinuses. Exacerbation of the disease in patients with CRSwNP leads to a significant decrease in the quality of life.
Aim. To study the bacterial component of the microbiota of nasal and paranasal mucosa in patients with CRSwNP during remission and exacerbation.
Materials and methods. 83 patients with CRSwNP were examined (44 patients in remission, 39 people in the period of exacerbation of the disease). A qualitative and quantitative analysis of bacterial component of the microbiota in all patients were carried out.
Results. No significant differences in the qualitative and quantitative composition of the nasal cavity microbiota during exacerbation and remission of inflammatory process were observed, as well as before and after treatment of the CRSwNP exacerbation. The quantitative assessment of the identified microorganisms in the vast majority of cases was within the normal range.
445-452
Nucleotide tetramers TCGA and CTAG: viral DNA and the genetic code (hypothesis)
Abstract
Introduction. The published and our own data show that CTAG and, to a lesser extent, TCGA tetra-nucleotides have significantly lower concentrations in frequency profiles (FPs) of herpesvirus DNAs compared to other complete, bilaterally symmetrical tetra-nucleotides.
The aim of the study is to present a comparative analysis of CTAG and TCGA tetra-nucleotide FPs in viral DNAs.
Materials and methods. We have analyzed FPs and other characteristics of the two above tetramers in DNAs of at least one species of viruses of each genus (or each subfamily, if the classification into genera was not available), complying with the size limit requirements (minimum 100,000 base pairs) — a total of more than 200 species of viruses. The analysis was performed using the GenBank database.
Results. Two groups of characteristics of TCGA and CTAG tetramers have been described. One of them covers the results of the FP analysis for these tetranucleotides in viral DNAs and shows that DNAs with GC:AT > 2 are characterized by nCGn FP symmetries while these symmetries are frequently distorted in nTAn FP due to CTAG underrepresentation. The other group of tetramer characteristics demonstrates differences in their FPs in complete viral DNAs and in their genomes (a coding part, which can reach 80% in some studied viruses, thus making the analysis of their DNAs more significant than the analysis of DNAs of cellular live forms) and suggests that these tetramers may have participated in the origin of the universal genetic code.
Discussion. Assumedly, the genetic code started evolving amid C+G prevailing in "pre-code" DNA polymers; then the initial code forms evolved further to their final structure where TCGA and CTAG tetramers hold a central position, encapsulating the previous stages of this evolution. The nCGn FP symmetries typical of the "complete" DNA of Herpes simplex viruses disappear in the sequence of the second codon letters of the genome of these viruses, implying that their functions differ from functions of other letters and emphasizing the reasonableness of presenting the genetic code as a calligram where the second line is not symmetrical.
478-493
The first case of detection of Listeria monocytogenes sequence types ST7, ST20, ST425 in wastewater during an investigation of water bodies in the Vologda region
Abstract
Introduction. Listeria monocytogenes is an important human pathogen causing various forms of listeriosis, including foodborne infections, meningitis, neonatal sepsis, and abortion. Listeria are common all over the world.
The purpose of the study was to conduct microbiological monitoring of L. monocytogenes in water reservoirs near livestock premises in the Vologda district of the Vologda region.
Materials and methods. Bacterial cultures were isolated using two methods, titration and filtration, followed by analysis using methods of conventional bacteriology, serotyping, and species identification by instrumental procedures such as whole genome sequencing, and bioinformatic analysis.
Results. Three isolates of L. monocytogenes and one isolate of Listeria innocua were isolated from 12 analyzed water samples (wastewater — 6, river water — 4, and storm water — 2 samples). whole genome sequencing of three L. monocytogenes strains attributed them to the evolutionary line II, and to three sequence types and two serogroups ST425(1/2a-3a), ST20(1/2a-3a), ST7 (4a-4c). The strains are shown to belong to multiple drug resistant ones conferring resistance to three functional groups of antibacterials such as tetracyclines, macrolides, and sulfonamides. Antibiotic resistance genes (fox, psp-like, lin,norB,sul), virulence Islands LIPI-1 and LIPI-2, and virulence genes inlABCJ, oatA, ami, gtcA, vip, and lisK in genomes of the strain were identified. Stress tolerance Island SSI-1 was identified in one strain.
Conclusions. The data obtained indicate contamination of water sources near the livestock premises with L. monocytogenes strains possessing high pathogenic potentiality for outbreaks of listeriosis in humans. This shows the necessity of careful monitoring of water sources for the presence of the causative agent of listeriosis as well as the implementing of anti-epidemic measures.
453-464
Molecular genetic characteristics of Vibrio cholerae nonO1/nonO139 strains isolated on the territory of Russian Federation from patients with otitis
Abstract
Introduction. In 2017–2020 for the first time in many years strains of Vibrio cholerae nonO1/nonO139 (NAGs) were isolated in Russia from patients with otitis.
Aim — bioinformatic analysis of whole genome sequences (WGSs) and sequences of individual genes of NAG strains - causative agents of otitis isolated in Russia.
Materials and methods. Analysis of WGSs of eight NAG clinical isolates obtained on the MiSeq Illumina platform was carried out using BioEdit, BLASTN, BLASTP, Vector NTI programs; antibiotic resistance was determined according to MUK 4.2.2495-09.
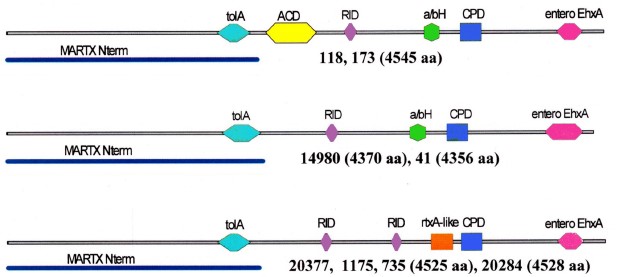
Results. The strains differed in SNP content, sets of determinants of pathogenicity/persistence factors and their alleles. All lacked CTX, preCTX, RS1 prophages, VPI pathogenicity island, thermostable toxin gene, mobile elements associated with antibiotic resistance, pandemicity island VSP-I; two strains contained VSP-II island. Genes of a number of proteases, cholix toxin, type 3 secretion system (T3SS) cluster and additional T6SS clusters formed different combinations. Products of the altered genes retained or lost their characteristic active domains. In the cytotoxin MARTX of 6 strains, the key ACD domain was absent; in 4 strains a new rtxA-like domain was revealed. Biofilm gene clusters varied in their structure. The presence of genes for antibiotic resistance did not always correlate with antibioticograms. All strains were susceptible to most antibiotics, but some showed resistance to 1–4 drugs.
Conclusion. All the studied strains — causative agents of otitis, in spite of revealed differences, have sufficient sets of determinants responsible for realization of pathogenic and persistent potential. Due to discrepancy between the genotypic and phenotypic characteristics of antibiotic resistance, one should rely mainly on the phenotype when choosing drugs for the etiotropic therapy of NAG infections. Emergence of patients with otitis caused by NAG-vibrios in Russia indicates the advisability of the inclusion of tests for their identification in the scheme of bacteriological analysis for extraintestinal infections and, in cases of their isolation, for prompt determination of sensitivity to antibiotics.
465-477
OBITUARIES
 494-495
494-495
ANNIVERSARIES
 496-497
496-497
CHRONICLE
498-498
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.