


Vol 99, No 3 (2022)

- Year: 2022
- Published: 28.07.2022
- Articles: 10
- URL: https://microbiol.crie.ru/jour/issue/view/50
Full Issue

ORIGINAL RESEARCHES
COVID-19: the evolution of the pandemic in Russia. Report I: manifestations of the COVID-19 epidemic process
Abstract
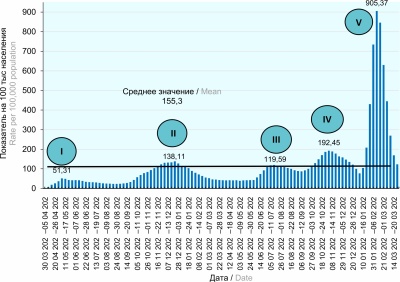
Background. The ongoing pandemic of a new coronavirus infection (COVID-19) determines the relevance of the analysis of epidemiological patterns of SARS-CoV-2 spread among the population of the Russian Federation.
Aim — study of the manifestations of the epidemic process of COVID-19 in the Russian Federation in 2020–2022.
Materials and methods. A retrospective epidemiological analysis of the incidence of COVID-19 in the Russian Federation was carried out from 03/30/2020 to 04/24/2022. The data from the Rospotrebnadzor report No. 970 “Information on cases of infectious diseases in persons with suspected new coronavirus infection”, information portal Stopcoronavirus.rf, etc. were used. The presence of SARS-CoV-2 RNA was confirmed by real-time RT-PCR.
Results and discussion. The analysis of the manifestations of the epidemic process of COVID-19 in the Russian Federation in 2020–2022 showed the presence of two stages which differed depending on the influence of the biological factor and the ongoing anti-epidemic measures. There was a pronounced trend in the development of the epidemic process, starting from megacities (Moscow, Moscow region and St. Petersburg), which are major transport hubs and centers of migration activity of the population, to the regions of the Russian Federation. The SARS-CoV-2 pathogenicity has been shown to decrease with each subsequent cycle of the rise in the incidence of COVID-19 against the background of the increased contagiousness of the virus.
Conclusion. As a result of the study, risk areas (megacities) and risk groups were identified.
 269-286
269-286



Clinical and epidemiological characteristics of hospitalized patients with COVID-19 during different pandemic periods in Moscow
Abstract
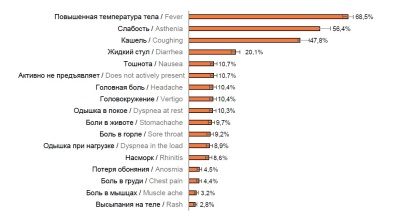
Background. The incidence of COVID-19 novel coronavirus infection has a wave-like pattern with surges in new cases followed by declines. Viral mutations, changes in viral properties, and new strains continue to emerge and are regularly reported.
The aim of the study is to present a comparative analysis of clinical and epidemiological characteristics of hospitalized patients with COVID-19 during different periods of the coronavirus infection pandemic in Moscow.
Materials and methods. A two-center, retrospective observational epidemiological study was performed using medical records of patients hospitalized with the confirmed diagnosis of COVID-19 in Moscow from March 2020 to March 2022 (34,354 patients).
Results. Within 2 years of the pandemic, there were significant differences in the age structure of hospitalized patients. During the early months (March–June 2020) of the pandemic, age groups of 18–45 and 46–65 yearolds accounted for higher percentages of hospitalizations. Later on (July 2020 – February 2021), the proportion of older age groups demonstrated an upward trend. From spring 2021 (the emergence of the SARS-CoV-2 delta strain) to March 2022 (dominance of the omicron strain), the proportion of hospitalized working-age adults increased once again.
The proportion of severe and critically severe cases among the patients hospitalized during different periods remained at steady levels: 7.7% (6.6–8.8%) and 5.5% (4.4–6.6%), respectively. The highest death rates were observed during the delta strain surge, while the lowest death rates were reported for the omicron strain. Throughout the pandemic, the older age and chronic diseases remained risk factors contributing to the severity of the disease and adverse outcomes.
Conclusion. The emergence of new variants of SARS-CoV-2 causing a shift of the need for hospitalization towards younger age groups, the persistent high rates of severe cases and death rates among people of retirement age are pressing for the unfailing readiness for implementing preventive and epidemic control measures focusing on the above groups of population.
287-299
Preclinical study of immunogenicity of adjuvanted quadrivalent subunit influenza vaccine
Abstract
Background. Preventive vaccination is a vitally important strategic aspect of protection of the population against severe effects of influenza epidemics. The priority attention is given to development of effective tetravalent vaccines containing antigens of two influenza A lineages (H1N1, H3N2) and two influenza B lineages (Victoria and Yamagata) in combination with immunoadjuvants.
The aim of the work was to conduct the preclinical study of the immunogenicity and protective efficacy of the innovative tetravalent subunit vaccine containing antigens of influenza A and B viruses as well as a corpuscular adjuvant.
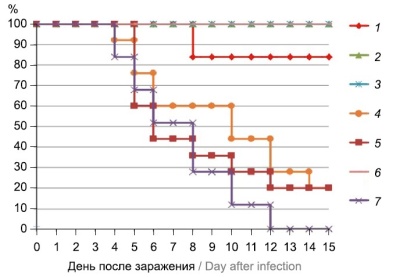
Materials and methods. The study was conducted using female BALB/c mice. The tetravalent vaccine and monovalent intermediate vaccines combined with a betulin adjuvant were injected intraperitoneally two times at a 14-day interval. The immunogenic activity was measured by the hemagglutination inhibition assay. The protective activity of the vaccine was assessed by changes in the viral load, body weight and survival rates using the mouse model of fatal influenza A H1N1 virus infection.
Results. The mice vaccinated with the adjuvanted quadrivalent subunit influenza vaccine produced antibodies against all four influenza viruses included in the vaccine; the mean antibody titers in the hemagglutination inhibition assay were above 1 : 40. The second-dose vaccination induced a significant increase in levels of antibodies against all four influenza viruses. The dose of the quadrivalent subunit adjuvanted vaccine containing 5 µg of each antigen and 200 µg of the adjuvant provided a 100% survival rate in mice and significantly decreased lung viral titers (more than 3 lg TCID50) in the mouse model of influenza pneumonia.
Conclusion. The quadrivalent subunit vaccine with the betulin-based corpuscular adjuvant demonstrates high immunogenicity in laboratory mice and provides protection against fatal pneumonia caused by the influenza A virus subtype H1N1.
300-308
Predicting incidence of Crimean-Congo hemorrhagic fever using satellite monitoring (remote sensing) data in the Stavropol Territory
Abstract
Introduction. With the epidemiological situation for Crimean-Congo hemorrhagic fever (CCHF) remaining tense in many countries worldwide, special attention should be focused on development and improvement of risk-based epidemiological prediction methods.
The aim of the study was to build a prediction model for CCHF incidence dynamics (based on the Stavropol Territory) using satellite monitoring (remote sensing) data.
Materials and methods. We analyzed the climate data obtained from the Space Research Institute of the Russian Academy of Sciences as well as the data of public statistics reports on CCHF incidence from 2005 to 2021. The prediction model incorporated the Bayes theorem and Wald sequential analysis. The information content of the factors was assessed using the Kullback method.
Results. Predictions for each of 26 districts were made stepwise (compared to threshold levels) to predict whether there will be at least one case of CCHF, whether the relative incidence per 100,000 population will exceed the median level (0.9 cases) or the average rate (3.5 cases) or the third quartile rate (4.7 cases). The highest values of information coefficients were obtained for soil temperature and moisture content (at depths of 10 and 40 cm), normalized relative vegetation index, relative humidity, maximum and average air temperature, relative air humidity. During the testing of the model in 2021, false-negative (erroneous) prediction was made for 2 districts.
Discussion. The model proved to be most efficient in prediction of occurrence or absence of cases. More accurate quantitative prediction may be difficult due to subjective factors (including misdiagnosing CCHF cases without hemorrhagic manifestations and administering treatment for other conditions with similar symptoms).
Conclusion. The tests of the model demonstrate its potential. The practical application of the prediction will make healthcare workers more alert when screening and detecting CCHF cases.
322-335
Whole genome sequencing of Acinetobacter baumannii strains isolated from hospital patients in the northern territories of the Tyumen region
Abstract
Introduction. is to The analysis of the genetic relatedness of isolates aiming to find the source of infection is an important task of nosocomial infection control. The most common causative agent of healthcare-associated infections is Acinetobacter baumannii.
Objective. To evaluate the results of whole genome sequencing of A. baumannii bacteria isolated from clinical samples of patients undergoing inpatient treatment in the northern territories of the Tyumen region.
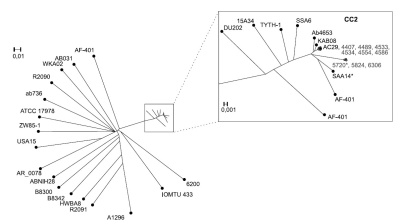
Materials and methods. Nine isolates of A. baumannii from the clinical material of patients were studied. Bacterial cultures were identified by mass spectrometry. Whole genome sequencing, multilocus sequence typing and search for markers of antibiotic resistance were performed.
Results. The studied strains belonged to sequence types ST2 and ST187, and to the international clonal complex CC2. All A. baumannii isolates were found to have beta-lactamase genes, as well as genes for resistance to aminoglycosides, to the MLS group of antibiotics, and to tetracyclines. The presence of a cluster of genes associated with virulence was detected: those responsible for the synthesis of acinetobactin and iron binding, surface antigen 1 and porin.
Conclusion. Based on data of a single nucleotide polymorphism (SNP) analysis, A. baumannii isolates from the clinical material of patients of healthcare institution #1 belong mainly to one bacterial strain. Isolates of A. baumannii from the clinical material of patients of healthcare institution #2 are closely related. The ability to distinguish clinical isolates of A. baumannii at the level of several SNPs per genome will improve the identification of the source of infection, and whole genome sequencing data can contribute to the rational prescription of antibiotic therapy and the correction of disinfection and antiseptic measures.
343-352
Characterization of Pseudomonas aeruginosa isolated from positive samples of hemocultures and cerebrospinal fluid of children
Abstract
Introduction. Infections of the bloodstream and central nervous system (CNS) caused by Pseudomonas aeruginosa are associated with a serious patient conditions and are often accompanied by high mortality.
Aim. Molecular genetic characterization of P. aeruginosa isolated from positive samples of blood cultures and cerebrospinal fluid of patients under 18 years of age from intensive care units of hospitals.
Materials and methods. We conducted a retrospective study of bacteremia and CNS infection cases associated with P. aeruginosa from 2014 to 2021. 24 clinical isolates of P. aeruginosa from positive blood cultures and CSF were analyzed. MICs of antibiotics were determined by serial microdilution in broth. Identification of the genes of carbapenemase was carried out using real-time PCR. Virulence genes were determined by PCR. Population diversity was assessed by MLST.
Results. More than 70% of isolates showed resistance to carbapenem antibiotics. The phenotype of multiple drug resistance had 25% of the isolates. Extreme resistance was shown by 54% of isolates. The detection rate of metallo-β-lactamases (MBL) was 54%. Based on PCR data, 33% of the strains were found to have the ExoU type, and 67% had the ExoS type. According to MLST, 16 genotypes were identified. The structure was dominated by two sequence types ST654 (29%) and ST235 (12.5%). The structure of patients was dominated by children with surgical pathology — 16 cases, and there were eight somatic patients. Fatal outcome was observed in 28% of cases with bacteremia and CNS infection associated with P. aeruginosa.
Conclusion. P. aeruginosa isolates from positive blood cultures and CSF samples are highly resistant to antibiotics; virulence genes were found in all isolates. Strains of high epidemic risk prevailed in the studied sample. More than a quarter of the described clinical cases had an unfavorable outcome.
309-321
Assessment of antibacterial activity of vinyltriphenylphosphonium hexabromoplatinate
Abstract
Introduction. One of the biological effects of platinum compounds is antibacterial action against Gram-positive and Gram-negative bacteria. Thus, some platinum compounds may inhibit the synthesis of DNA, RNA and proteins in Escherichia coli cells.
Aim — to study the antibacterial effect of vinyltriphenylphosphonium hexabromoplatinate (VH) against E. coli and Staphylococcus aureus bacteria.
Materials and methods. The antibacterial effect of VH was studied by the quantitation of the grown colonies of E. coli strain ATCC 25922 and S. aureus strain ATCC 6538 on a nutrient medium in test (suspension of microorganisms, solution of the test substance) and control (suspension of microorganisms). The significance of differences between the outcomes in test and control groups was estimated by two-sided Fisher's exact test.
Results. The minimum inhibitory concentration of VH solution for E. coli strain ATCC 25922 was 14,0625 µg/ml, for S. aureus strain ATCC 6538 — 225 µg/ml.
Conclusion. VH exhibits a more pronounced antibacterial effect against E. coli compared to S. aureus — the minimum inhibitory concentration of VH observed for E. coli (14,0625 µg/ml, 11,17 µM) is comparable to the effective concentrations of platinum antitumor compounds — therefore, further studies with this bacterium, including in vivo studies, are promising.
336-342
REVIEWS
Genetic polymorphism associated with cervical cancer: a systematic review
Abstract
Introduction. Cervical cancer (CC) is one of the most common cancers in women. The CC etiological agent is the high-risk oncogenic human papillomavirus. In the meantime, not all women infected with this virus can develop cancer, thus suggesting that there is genetic predisposition to CC.
The aim of the study was to analyze information about single nucleotide polymorphisms associated with the CC risk.
Materials and methods. The performed search was focused on genome-wide association studies (GWAS) and meta-analyses conducted over the last 10 years and addressing the genetic risk of CC in the Caucasian population.
Results. The most significant associations with CC were found in the following single nucleotide polymorphisms. Based on the GWAS data, they involve risk alleles rs138446575-T (OR = 2.39) TTC34; rs73728618-T (OR = 1.48) HLA-DQA1; rs3130196-C (OR = 1.4) HLA-DPB1; rs2516448-T (OR = 1.39 and 1.44) MICA and protective alleles rs9271898-A (OR = 0.64) and 9272143-C (OR = 0.65) between HLA-DRB1 and HLA-DQA1, rs55986091-A HLADQB1 (OR = 0.66). Based on the meta-analysis data, they involve genotype rs4646903-СС (OR = 4.65) CYP1A1 and protective alleles rs1801133-T (OR = 0.77) MTHFR, rs2333227-AA (OR = 0.57) MPO.
Conclusion. The obtained data are critically important for development of laboratory techniques and reagent kits allowing for a personalized approach to identification of risk groups, which could benefit from compulsory vaccination and screening for pre-cancers of the cervix.
353-361
Determinants of resistance of Francisella tularensis to environmental stress
Abstract
The review summarizes current literature data on the main structures and components of the tularemia microbe responsible for adaptation to the warm-blooded host macroorganism (susceptible animals, humans). According to scientific data, the successful survival of Francisella tularensis under stress conditions requires the interaction of all cellular structures of the microbe. Despite active research carried out in the field of studying the determinants and mechanisms of F. tularensis resistance, the reason for the high adaptive capacity with low variability of the tularemia pathogen has not been established. These studies are important for understanding the mechanisms of persistence and virulence of F. tularensis, as well as for further development of vaccines and diagnostic tests.
362-371
ANNIVERSARIES
 372-373
372-373
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.