


Vol 97, No 6 (2020)

- Year: 2020
- Published: 20.01.2021
- Articles: 16
- URL: https://microbiol.crie.ru/jour/issue/view/41
Full Issue

ORIGINAL RESEARCHES
Genetic variability of SARS-CoV-2 in biological samples from patients in Moscow
Abstract
Currently, a lot of attention is given to SARS-CoV-2 subpopulations and their coexistence with different genomic variants within the same patient. In this study, we performed next-generation whole-genome sequencing and assembly of viruses from samples representing swabs or autopsy specimens obtained from patients diagnosed with СOVID-19, which were initially confirmed by the real-time polymerase chain reaction (Ct = 10.4–19.8). Samples were prepared for sequencing by using the SCV-2000bp protocol. The obtained data were checked for presence of more than one SARS-CoV-2 genetic variants in a sample. Variants of nucleotide substitutions, coverage for each variant, and location of the variable position in the reference genome were detected with tools incorporated in the CLC Genomics Workbench program. In our search for variable nucleotide positions, we assumed that the sample had two genetic variants (not more); the threshold value ≥ 90% was set for probability of the identified variant. Variants represented by less than 20% of the reads in the total coverage were not taken into consideration. The obtained results showed that 5 samples had variability, i.e. they had several genetic variants of SARS-CoV-2. In 4 samples, both of the detected genomic variants differed only in one nucleotide position. The fifth sample demonstrated more substantial differences: a total of 3 variable positions and one three-nucleotide deletion. Our study shows that different genetic variants of SARS-CoV-2 can coexist within the same patient.
 511-517
511-517



Distribution of SARS-CоV-2 seroprevalence among residents of the Republic of Tatarstan during the COVID-19 epidemic period
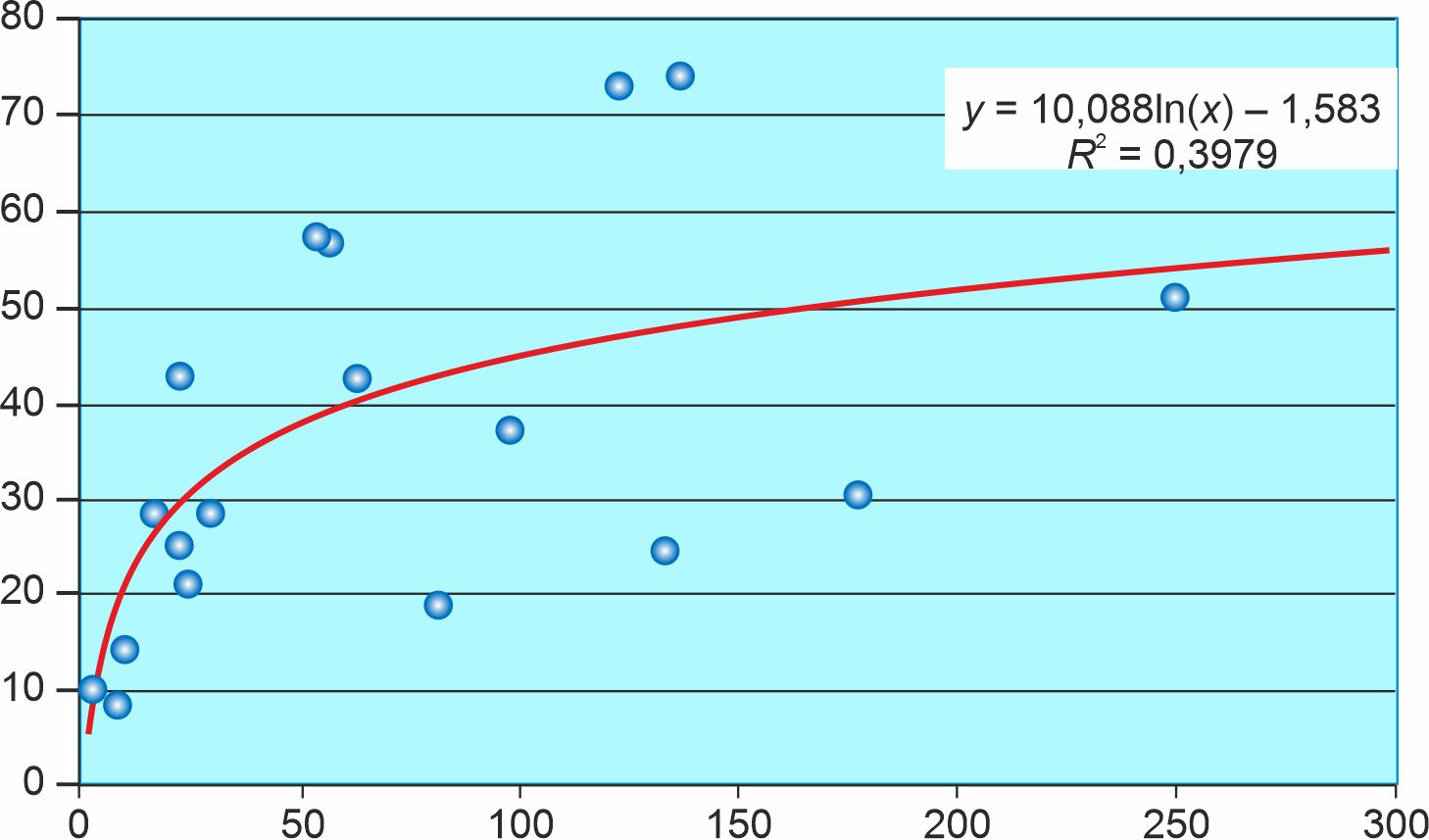
Abstract
In late 2019, there were reports of an outbreak of infection caused by a new strain of beta coronavirus SARSCoV-2, the WHO identified the disease as coronavirus disease 2019 (COVID-19). In Tatarstan, the first case of COVID-19 was diagnosed on March 16, 2020, it was an imported case from France. The period of increase in the incidence lasted during the 12th to the 19th week, when the highest rate was recorded, amounting to 16.7 per 100 thousand population. Subsequently, a statistically significant decrease in the incidence was noted. Seroprevalence study was conducted at week 27 (8th week of decline of morbidity).
The purpose of the seroepidemiological study was to measure the level and to identify the structure of herd immunity against the SARS-CoV-2 virus among the population of the Republic of Tatarstan during the rapid spread of the COVID-19 outbreak.
Materials and methods. The selection of volunteers for the study was carried out by the method of questionnaires and randomization by random sampling. The exclusion criterion was active COVID-19 infection at the time of the survey. 2,946 people were examined for the presence of specific antibodies to SARS-CoV-2. The age of the surveyed volunteers ranged from 1 year to 70 years and older.
Results. The results of the study showed that in the Republic of Tatarstan during the period of COVID-19 incidence, there was a moderate seroprevalence to SARS-CoV-2, which amounted to 31.3%, against the background of a high frequency (94.5%) of asymptomatic infection in seropositive individuals who did not have a history of past COVID-19 disease, positive PCR result and ARVI symptoms on the day of the examination. The maximum indicators of herd immunity were established in children aged 7–13 years (42.0%), children 14–17 years old (40.3%), with a simultaneous decrease in seroprevalence in persons aged 70 and older (24.0%). In different regions of the Republic of Tatarstan, there was a wide variation in seropositivity results from the minimum in the Zainsky district (8.6%) to the maximum in the Arsky district (74.3%). In 21 out of 38 surveyed districts, the results were unrepresentative due to the small sample size. In COVID-19 convalescents, antibodies are produced in 83.3% of cases. In persons with a positive result of the PCR analysis carried out earlier, antibodies were detected in 100% of cases. Among the volunteers who had contact with patients with COVID-19, the proportion of seropositive is 37%
Conclusion. The dynamics of seroprevalence among the population of the Republic of Tatarstan can be qualified as positive, the results obtained can be used to develop a forecast for the development of the epidemiological situation, as well as to plan measures for specific and non-specific prevention of COVID-19.
518-528
Sensitivity of biofilms of vaccine and freshly isolated Bordetella pertussis strains to antibiotics
Abstract
Aim. To study the sensitivity of biofilms of vaccine and freshly isolated strains of Bordetella pertussis to antibiotics.
Materials and methods. Vaccine and freshly isolated strains of B. pertussis were used. Cultures of strains grown on dense nutrient medium were used as inoculate for biofilms production. The intensity of biofilm formation in round-bottomed polystyrene 96-well plates was estimated by staining with 0.1% gentian-violet solution. The following antibiotics were used in experiments: penicillins (ampicillin), cephalosporins (ceftriaxone), aminoglycosides (gentamicin), macrolides (erythromycin).
Results. The highest resistance to antibiotics was demonstrated by the vaccine strain No. 305 and freshly isolated strain No. 211, sensitive only to erythromycin. Vaccine strain No. 703 was sensitive to gentamicin and ampicillin and showed resistance to erythromycin and ceftriaxone. Vaccine strain No. 475 was sensitive to all tested antibiotics. The Tohama 1 strain was resistant to ampicillin and sensitive to other antibiotics. Freshly isolated strains No. 178 and No. 162 were resistant to ceftriaxone and sensitive to gentamicin, erythromycin and penicillin. Minimal inhibitory concentrations of tested antibiotics ranged from 0.2 μg/ml to 5.0 μg/ml.
Conclusion. These data indicate the heterogeneity of vaccine and freshly isolated strains of B. pertussis in sensitivity to antibiotics. The greatest activity was shown by erythromycin, which suppressed the growth of biofilms of 6 out of 7 strains. The least effective was ceftriaxone, which suppressed the growth of biofilms of only 2 strains.
529-534
Epidemiology and molecular genetic characteristics of Lyme borreliosis pathogens circulating in tick’s population in the Almaty oblast of the Republic of Kazakhstan
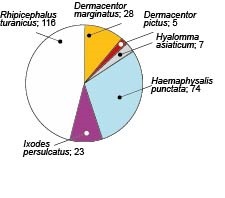
Abstract
Background. Information on the geographical distribution of different species of the Borrelia burgdorferi sensu lato (B. burgdorferi s.l.) complex is of great epidemiological importance, since different genospecies are associated with certain clinical manifestations of Lyme borreliosis. Although Almaty region of the Republic of Kazakhstan is considered to be endemic for tick-borne borreliosis, there is still no accurate data on the level of borrelia infection in ticks in the region, including information on the genotypes of circulating borrelia.
The aim of this work was to study ticks collected from humans in the Almaty region of the Republic of Kazakhstan in 2018.
Materials and methods. Ticks were tested for the presence of B. burgdorferi s.l. DNA, genotyping of the identified borrelia was done by sequencing of the fragment of 16S rRNA gene. The analysis of epidemiological data on the incidence of Lyme borreliosis in the Almaty region in 2013–2018 was performed.
Results. Rhipicephalus turanicus (116/253), Haemaphysalis punctata (74/253), Dermacentor marginatus (28/253), and Ixodes persulcatus (23/253) were the predominant species of ticks taken from humans. The prevalence of B. burgdorferi s.l. infection in I. persulcatus ticks was 39.13% (9/23) It should be noted that the DNA of B. burgdorferi s.l. was also detected in single individuals of D. marginatus, H. punctata, and R. turanicus, although these species are not considered as competent B. burgdorferi s.l. vectors.
Conclusion. As a result of sequencing of the positive homogenates of ticks, two genotypes of B. burgdorferi s.l. were identified: B. afzelii and B. garinii and/or B. bavariensis. Thus, at least two genospecies, B. afzelii and B. garinii and/or B. bavariensis, circulate in the territory of the Almaty region.
535-545
Analysis of sporadic cases of invasive listeriosis in a metropolis
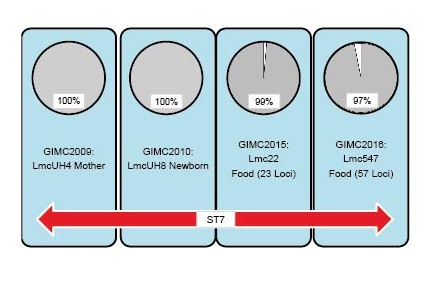
Abstract
Introduction. Listeriosis is a foodborne infection, especially dangerous for people in at-risk groups. Susceptibility to listeria infection is determined by a complex of reasons: environmental factors, host immune status, and pathogen virulence. The susceptibility to listeriosis can also be aggravated by previous infections, especially viral infections, which demonstrate a steadily increasing number of identified pathogens.
The aim of our study was to present molecular and genetic characterization of pathogens causing sporadic invasive listeriosis in a megalopolis, primarily during the peak of influenza and ARVI incidence.
Materials and methods. Listeria monocytogenes isolates were collected from 18 hospitalized patients at hospitals in Moscow, from November 2018 to October 2019. The first comparison group was represented by isolates from food products and fish preserves. The second comparison group included previously examined environmental isolates. The clinical isolates were examined by using multilocus sequence typing techniques, including the standard MLST scheme extended by loci of internalin genes. Isolates of the autochthonous genotype (ST7) were compared through whole-genome sequencing and subsequent analysis of the core genome (cgMLST).
Results. In cases of invasive listeriosis, 44% of isolates were isolated from patients with listeriosis; 27% of isolates were obtained from patients with meningitis. L. monocytogenes of phylogenetic lineage II prevailed in these groups of cases that occurred when the epidemic threshold for influenza was crossed during the 2018/2019 season. Listeria pneumonia identified in the senior age group occurred during the season of autumn ARVI and was primarily caused by L. monocytogenes of phylogenetic lineage I. The examination of genomes of ST7 isolates demonstrated identity between the core genomes of bacteria isolated from the mother-infant pair. Out of ST7 food isolates most closely related to the clinical ones was the isolate from meat (23 locus differences, the common deletion in the MFS transporter locus). Analyzing invasive listeriosis, the comparison between the list of the identified genotypes and the data from European countries showed that each country had its own specific range of genotypes, though ST7 was detected in all the examined samples.
Conclusions. Along with the monitoring of food manufacturing and storage, timely vaccination against seasonal respiratory infections and use of personal protective equipment in public spaces can reduce the risk of listeriosis incidence in at-risk groups.
546-555
The study of neuraminidase immunity in protection against secondary bacterial pneumonia induced by S. aureus after influenza infection in mice
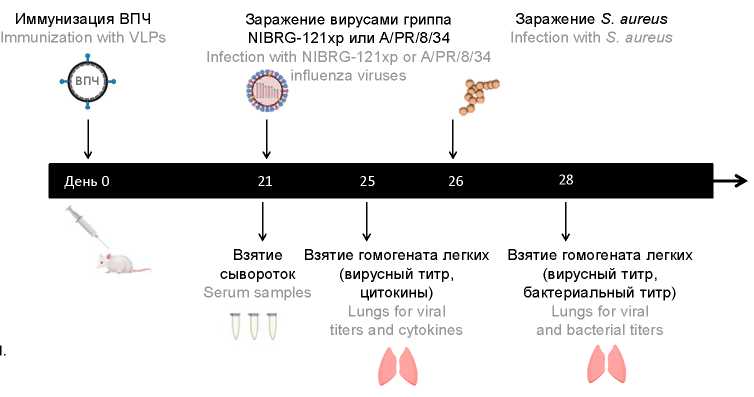
Abstract
Introduction. Pneumonia often occurs secondary to influenza infection and accounts for a large proportion of the morbidity and mortality associated with seasonal and pandemic influenza outbreaks. We previously have shown that vaccination with Virus-like particles (VLPs) containing hemagglutinin (HA) of influenza virus reduces mortality caused by bacterial infections after an influenza infections in mice.
The aim of this work is to study whether this protective effect may be potentiated by supplementing the HA preparation with the influenza neuraminidase (NA).
Materials and methods. We studied the effect of Gag-VLPs with the influenza HA or NA from А/PR/8/34 alone or in combination, in a lethal BALB/c mouse model of S. aureus infection after vaccine-matched or mismatched influenza virus challenge.
Results. A cocktail of HA-Gag and NA-Gag-VLPs fully protected from weight loss, mortality and viral replication and significantly reduced the bacterial burden in the lungs of А/PR/8/34 infected animals. Immunization with this cocktail HA-Gag-VLPs 100 ng + NA-Gag-VLPs 20 ng also protected 60% of animals from mortality associated with secondary bacterial S. aureus infection following a heterologous H1N1 influenza virus challenge, and led to the significant protection from weight loss and pulmonary pathogen replication even in the absence of HA-inhibition and NA-inhibition antibodies.
Conclusion. Our results indicate that influenza vaccination may improve the outcome of a secondary bacterial pneumonia induced by S. aureus after influenza even when the virus is antigenically different from the vaccine strain. At the same time, in our model, the significance of the immunity to influenza virus HA was prevalent.
564-577
Proteomic analysis of typical and genetically altered strains of Vibrio cholerae serogroup O1, biovar El Tor
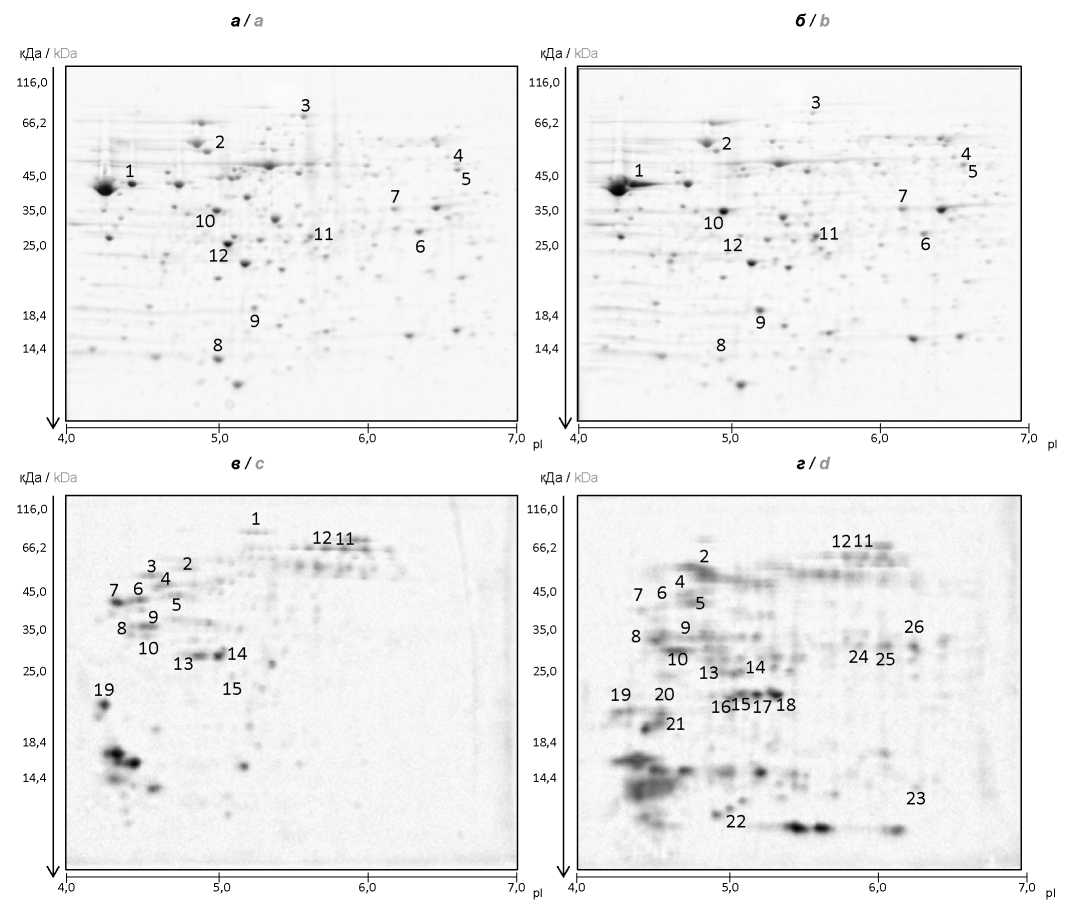
Abstract
Objective — comparative study of protein expression in typical and genetically altered Vibrio cholerae strains of O1 serogroup, biovar El Tor by means of proteomic analysis.
Materials and methods. Clinical V. cholerae strains — typical strain, M106 (Astrakhan, 1970) and genetically altered one, M1509 (Moscow, 2012) — were used as model ones. Strains were cultivated in LB broth (pH7.2). Then, cell and exoprotein lysate fractions were obtained and investigated in 2D electrophoresis. Different protein stains were examined using mass spectrometry. Survivability of V. cholerae strains under osmotic and oxidative stresses was studied during incubation of the strains in 3 M NaCl solution or 20 mM H2O2 solution.
Results and discussion. When analyzing cell lysates, significant differences in protein expression with known function between studied strains were not detected. The great majority of identified proteins in the lysates is functionally associated with carbohydrate metabolism, amino acid metabolism, and energy processes in a cell. At the same time, exoprotein fraction of M1509 genovariant contained increased amount of proteins (peroxidase, superoxide dismutase, thioredoxin, outer membrane proteins OmpW, OmpT) protecting the cells of cholera vibrio from effect of stress factors of the environment. Further study of the resistance to osmotic and oxidative stresses revealed better survivability in the genovariant when exposed to the stated factors.
Conclusion. The data of proteomic analysis of the typical and genetically altered V. cholera strains, biovar El Tor, testify to high levels of expression of the proteins that provide for vibrio resistance to the effect of environmental stress factors in genovariants, which is possibly one of the causes of their wide dissemination. In addition, the results obtained will allow for identification of new biomarkers which can be used for differentiation of typical strains and genovariants of V. cholerae, biovar El Tor.
578-586
Improvement of the technique of SNP-typing of Vibrio cholerae strains on the basis of the analysis of the primary data of whole genome sequencing
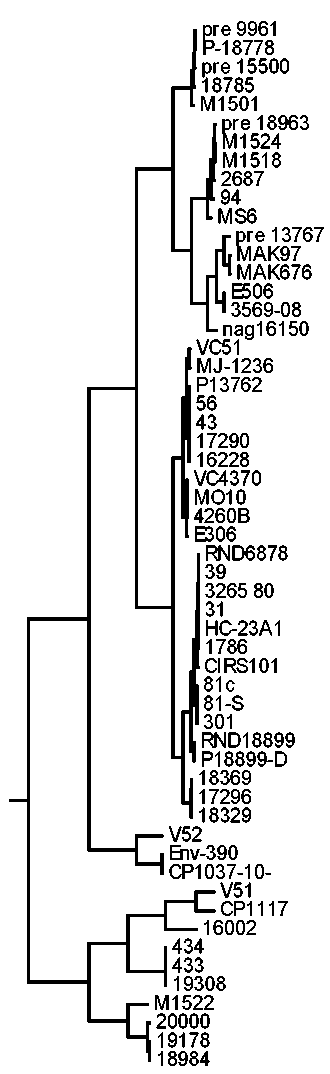
Abstract
Aim. To improve the method of the quality assessment of single nucleotide polymorphisms, which are used for SNP-typing, based on the analysis of their distribution in the primary data of whole genome sequencing (reads).
Materials and methods. Data of the whole genome sequencing of 56 Vibrio cholerae strains obtained using different types of sequencers were used. The software was developed using Java programming language. Cluster analysis and construction of the dendrogram were performed with the author's software using the UPGMA method.
Results and discussion. The «instability» of detection the number of SNP in the genome of cholera causative agent was shown. The method of selection of the SNP list for phylogenetic analysis based on the analysis of the primary data of whole genome sequencing (reads), has been developed. The method of using «control genomes» for cluster analysis of whole genome sequencing data has been proposed.
Conclusion. The list of 3198 «stable SNP» for phylogenetic analysis has been composed. Genetic affinity between the non-toxigenic strains that contain the tcpA gene (ctxAB–tcpA+) and preCTX-strains of V. cholerae was shown.
587-593
The relatedness of Klebsiella pneumoniae strains based on phylogenetic analysis of uge and fim genes
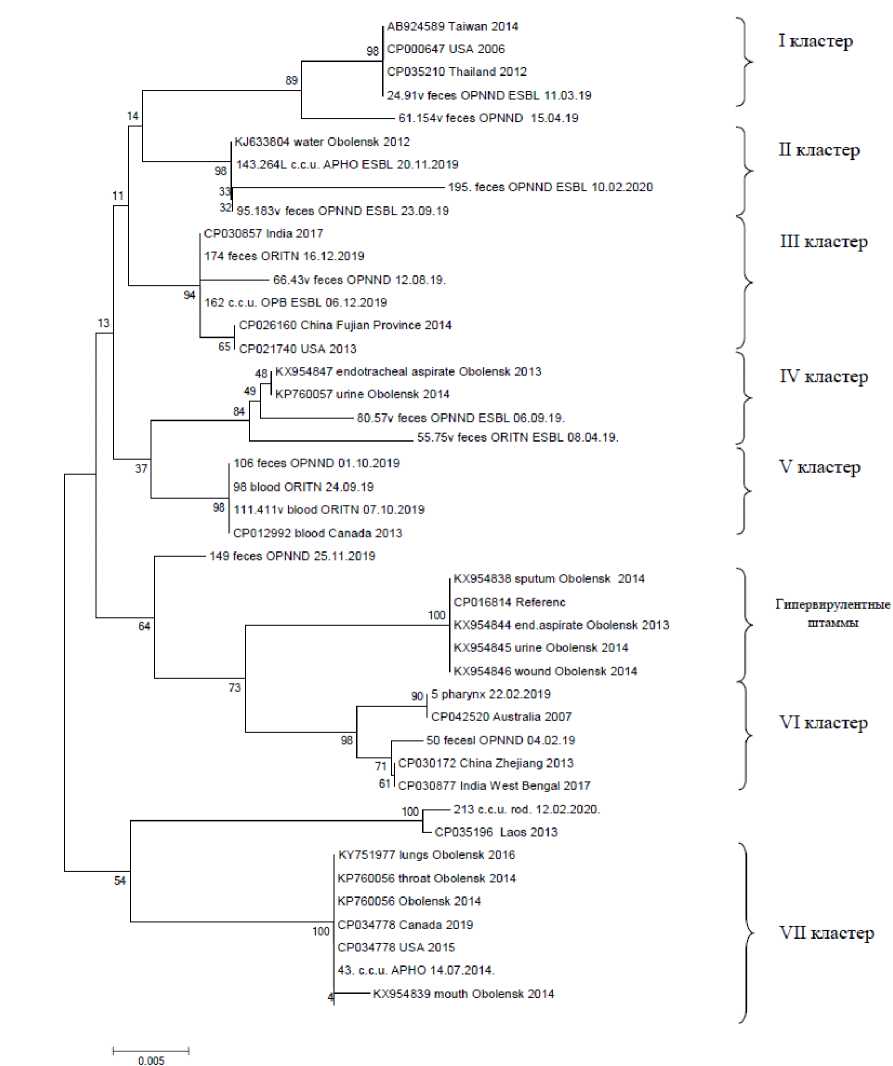
Abstract
Introduction. Currently, there is insufficient data on the prevalence of Klebsiella pneumoniae strains with virulence factors genes uge and fim among women and newborns. This indicates the need for a study of the prevalence of K. pneumoniae (uge+, fim+) and the degree of heterogeneity of the bacterial population isolated from children and adults.
The aim of the study was to perform a phylogenetic analysis of the uge and fim genes of the K. pneumoniae strains.
Materials and methods. Total 65 strains of K. pneumoniae isolated from samples of feces, blood, urine, placenta, cervical canal, pharynx, suture of 39 newborns and 24 women were studied. Two blood cultures were obtained from one patient with an interval of two weeks, and two isolates were obtained from the separated cervical canal and suture of one patient. The presence of genes was detected by PCR, nucleotide sequences of the genes were determined by Sanger sequencing.
Results. The frequency of detection of the uge gene was 53.8% (35 of 65), fim gene — 23.1% (15 of 65), which indicates a higher prevalence of uge gene strains compared to fim (p < 0.001). The phylogenetic analysis of 18 nucleotide sequences of the uge gene and 4 of the fim gene demonstrated that the strains were distributed in 7 and 4 clusters, respectively. It was established that for, there are No clear clustering by time and place of isolation, patient age, and type of biological material was observed for both uge and fim genes.
Discussion. The results of phylogenetic analysis demonstrate the genetic heterogeneity of the studied population of K. pneumoniae, which is confirmed by the wide geography and time variations in detection of the most genetically close bacterial isolates.
556-563
REVIEWS
The role of membrane phospholipids in the implementation of protective strategies of bacteria
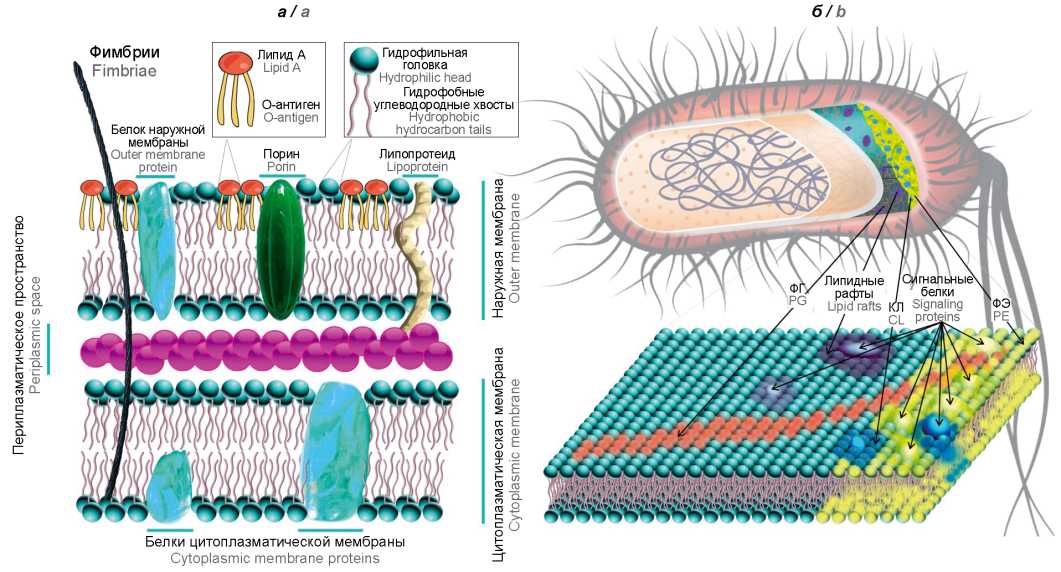
Abstract
To maintain viability under stressful conditions of existence and the implementation of protective strategies, bacteria must receive signals and respond quickly to extreme changes in environmental parameters. The results of recent experimental studies complement the paradigm that has dominated since the 1970s on the predominant role of phospholipids (PL) as molecular building blocks in the formation of the cell wall of bacteria. Specific transformations of these lipid domains have shown to have a significant effect on the shape and function of cells, membrane remodeling, and the ability of bacteria to adapt to environmental stresses. The physiological role of bacterial PLs is pleiotropic and determines both cell integrity and cell function. In addition to the key structural role of membrane PL in the cell, their intermediate metabolites are able to act as secondary messengers and perform important signaling and regulatory functions. Modern studies of the mechanisms of detection and integration of signals from the environment that cause stationary-dynamic changes in phospholipid homeostasis and form pleiotropic resistant cellular bacterial phenotypes are of fundamental and practical interest. PL homeostasis was proved to be crucial for the pathogenesis of bacterial infections and is necessary not only to maintain the viability of bacteria, but also to ensure their growth during infection. The suppression of the biosynthesis of these macromolecules reduces the viability of bacteria. In recent decades, one of the main advances in the concept of "liquid mosaic" model of biological membranes has been the understanding of their domain structure. This discovery is of fundamental and practical interest, since phospholipid domains are a promising target for modern antimicrobial strategies. The aim of this review is to summarize modern ideas about the structural, metabolic and signaling role of membrane PL in the implementation of the protective mechanisms of bacteria and maintaining their viability in adverse environmental conditions.
594-603
SHORT COMMUNICATIONS
Factors affecting mortality for the novel coronavirus infection in different regions of the Russian Federation
Abstract
Background. The influence of such factors as population density, practices for testing for the SARS-CoV-2 (combined with quarantine/self-isolation for infected individuals and their contacts) and ambient temperature on the spread of the novel coronavirus infection and related mortality in the 85 different regions of the Russian Federation isn’t well characterized.
Materials and methods. Population density in the different regions of the Russian Federation is measured as the number of persons per square kilometer of settled areas; ambient temperature is measured as the mean for January and July values; practices for testing for SARS-CoV-2 are characterized via case-fatality rates (the percent of deaths among cases with known outcome (recovered + fatal)) — under more active testing for SARSCoV-2, greater numbers of mild/moderate cases of infection are detected, resulting in the decline in case-fatality rates, i.e. the intensity of testing is inversely proportional to the case-fatality rate.
Results. The correlation between population density and rates of mortality for COVID-19 per 100,000 persons on November 22, 2020 in the 85 different regions of the Russian Federation is 0.53 (0.36; 0.67); the correlation between case-fatality rates and rates of mortality for COVID-19 per 100,000 persons on Nov. 22, 2020 in the different regions of the Russian Federation is 0.62 (0.47; 0.74). Results of the linear regression suggest a positive association between population density, as well as case-fatality rates and rates of mortality for COVID-19 in the different regions of Russia, and a negative association between ambient temperature and rates of mortality for the novel coronavirus infection.
Conclusions. Lower population density, more active testing for SARS-CoV-2 and higher ambient temperature are associated with lower rates of mortality for COVID-19. In particular, additional measures should be implemented towards testing of different categories of individuals for SARS-CoV-2, including those seeking testing on their own initiative, those seeking medical help with respiratory symptoms, and contacts of confirmed COVID-19 cases.
604-607
CHRONICLE
 608-609
608-609
 610-612
610-612
 613-614
613-614
OBITUARIES
 615-616
617-620
615-616
617-620
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.