


Том 99, № 4 (2022)

- Год: 2022
- Выпуск опубликован: 27.09.2022
- Статей: 13
- URL: https://microbiol.crie.ru/jour/issue/view/51
Весь выпуск

ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
COVID-19: эволюция пандемии в России. Сообщение II: динамика циркуляции геновариантов вируса SARS-CoV-2
Аннотация
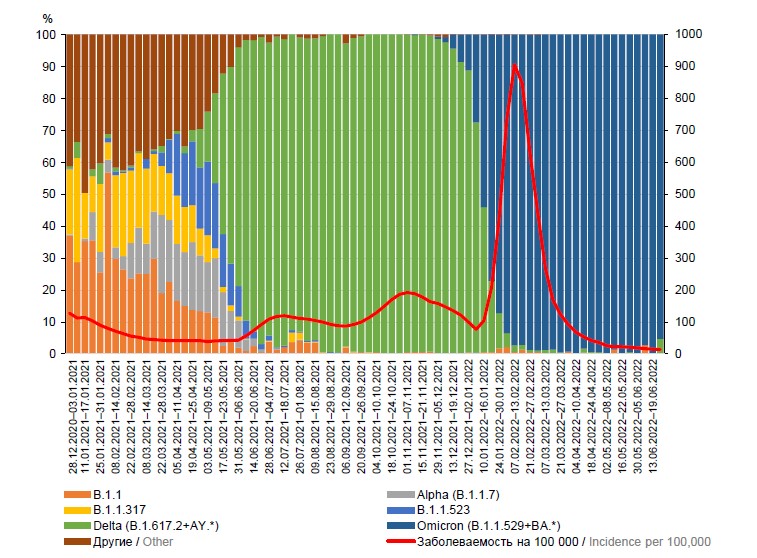
Актуальность. Продолжающаяся пандемия новой коронавирусной инфекции (COVID-19) определяет актуальность проведения молекулярного-генетического мониторинга распространения SARS-CoV-2 среди населения Российской Федерации.
Цель работы — анализ динамики циркуляции геновариантов вируса SARS-CoV-2 на территории России.
Материалы и методы. Проведён анализ динамики циркуляции геновариантов вируса SARS-CoV-2 с 28.12.2020 по 26.06.2022 на территории России. Использованы материалы отчёта Роспотребнадзора № 970 «Информация о случаях инфекционных заболеваний у лиц с подозрением на новую коронавирусную инфекцию», Российской платформы агрегации информации о геномах вирусов (VGARus). Наличие РНК SARS-CoV-2 было подтверждено методом полимеразной цепной реакции в режиме реального времени с обратной транскрипцией. Для проведения амплификации фрагментов генома и последующего секвенирования использовались разработанные в ЦНИИ Эпидемиологии праймерные панели.
Результаты и обсуждение. С помощью российской платформы VGARus, развёрнутой на базе ЦНИИ Эпидемиологии, получены данные о мутационной изменчивости SARS-CoV-2. Мониторинг циркуляции геновариантов SARS-CoV-2 на территории России с 28.12.2020 по 26.06.2022 выявил доминирование геновариантов Delta и Omicron на различных этапах эпидемии.
Заключение. Данные молекулярно-генетических исследований являются важнейшим компонентом эпидемиологического надзора для принятия управленческих решений по предотвращению дальнейшего распространения SARS-CoV-2 и формируют основу для создания новых вакцинных препаратов.
 381-396
381-396



Особенности распространения новой коронавирусной инфекции на территории муниципальных образований Ростовской области
Аннотация
Введение. С момента регистрации первых случаев COVID-19 вследствие высокой миграционной активности населения новая коронавирусная инфекция получила пандемическое распространение по всему миру, включая Россию.
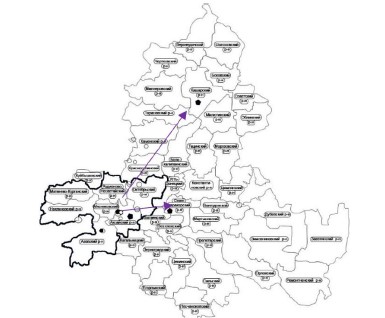
Цель работы — установить особенности распространения новой коронавирусной инфекции на территории Ростовской области.
Материалы и методы. На основании анонимизированных данных Управления Роспотребнадзора по Ростовской области проведен анализ 81 случая завоза новой коронавирусной инфекции лицами, прибывшими в Ростовскую область из-за рубежа или других субъектов РФ. Изучена динамика распространения COVID-19 по административным территориям Ростовской области. В работе использованы данные результатов полногеномного секвенирования (n = 155), проведённого на базе Ростовского-на-Дону противочумного института.
Результаты. В период с 21.03.2020 по 28.03.2020 в Ростовской области зарегистрированы завозные случаи как из-за рубежа, так и из других субъектов РФ, преимущественно на территории Ростовской городской агломерации. Вектор распространения болезни направлен от административного центра области к периферии. Появление новой генетической линии В.617.2 (Delta), вероятно, привело к значительному росту заболеваемости на территории Ростовской области.
Выводы. Распространению новой коронавирусной инфекции в Ростовской области способствовала реализация одного из основных социальных факторов эпидемиологического риска — миграция населения, что привело к завозу инфекции в административный центр субъекта — Ростов-на-Дону. С учётом ряда особенностей Ростовской области, наибольшая доля заболевших COVID-19 зафиксирована в Ростовской городской агломерации. На фоне доминирования геноварианта Delta на территории Ростовской области отмечалась тенденция к росту числа заболевших.
410-419
Биологическая характеристика холодоадаптированных вариантов коронавируса SARS-CoV-2
Аннотация
Введение. В связи с появлением новых эпидемиологически значимых вариантов SARS-CoV-2 актуальной является разработка живой вакцины, способной обеспечить защиту против широкого спектра антигенных вариантов вируса.
Целью исследования являлись получение и биологическая характеристика аттенуированных путём холодовой адаптации вариантов SARS-CoV-2.
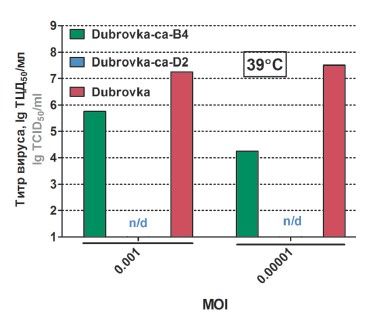
Материалы и методы. Лабораторный штамм SARS-CoV-2 Dubrovka и его варианты культивировали в клетках Vero и Calu-3. Количественное определение вируса проводили путём титрования в клетках Vero и методом полимеразной цепной реакции с обратной транскрипцией в режиме реального времени. Вирионы SARS-CoV-2 характеризовали методом трансмиссионной электронной микроскопии. Геномные последовательности вируса определяли методом нанопорового секвенирования. Аттенуационный (att) фенотип вариантов SARS-CoV-2 определяли на животной модели COVID-19 на сирийских хомяках.
Результаты. В результате длительного пассирования штамма Dubrovka в культуре клеток Vero при постепенно понижаемой до 23ºС температуре и последующего клонирования получены холодоадаптированные (ca, cold-adapted) варианты SARS-CoV-2 Dubrovka-са-B4 и Dubrovka-са-D2. В геномах ca-вариантов обнаружено до 20 нуклеотидных и 18 аминокислотных замен. Са-варианты, в отличие от родоначального штамма Dubrovka, эффективно размножались при 23ºС, а вариант Dubrovka-са-D2 имел температурочувствительный (ts) фенотип (не размножался при температуре 39ºС). Са-варианты вируса плохо размножались при температуре 37ºС в культуре клеток лёгких человека Calu-3, что, наряду с ts-фенотипом, может быть маркером аттенуации вируса по отношению к человеку. При интраназальном заражении сирийских хомяков ca-варианты вируса проявили аттенуационный фенотип — не приводили к снижению аппетита, вялости, сонливости, не замедляли прироста массы тела, значительно медленнее размножались в лёгких и мозге по сравнению с вирулентным штаммом Dubrovka.
Заключение. Полученные в настоящей работе аттенуированные са-варианты SARS-CoV-2 Dubrovkaса-B4 и Dubrovka-са-D2 представляют интерес для дальнейшего исследования в качестве кандидатных вакцинных штаммов для создания живой аттенуированной вакцины против COVID-19.
397-409
Исследование in vitro механизмов взаимодействия грибов Candida albicans с Klebsiella pneumoniae и Enterococcus faecalis, выделенных из кишечного микробиома ВИЧ-инфицированных пациентов
Аннотация
Цель: определение in vitro мишеней для факторов антагонизма клебсиелл и энтерококков у грибов Candida albicans, выделенных из кишечного микробиома ВИЧ-инфицированных пациентов.
Материалы и методы. В экспериментах использованы 38 штаммов грибов Candida albicans, 28 штаммов Klebsiella pneumoniae и 30 штаммов Enterococcus faecalis, изолированных из кишечного микробиома 89 ВИЧ-инфицированных детей. Средний возраст пациентов составил 24 ± 2 мес, мальчиков было 49 (55%), девочек — 40 (45%). Микроорганизмы выделяли из кишечного биотопа с использованием селективных питательных сред HiChrome Candida Agar, HiChrome Klebsiella Selective Agar Base, Энтерококкагар; проводили видовую идентификацию. В модельных экспериментах изучена антикаталазная активность экзометаболитов E. faecalis и влияние K. pneumoniae на морфологическую трансформацию грибов C. albicans.
Результаты. Клебсиеллы на 58,7% снижают интенсивность образования ростовых трубок у C. albicans (p < 0,01). При совместном культивировании 12,3% дрожжевых клеток дают ростовые трубки, тогда как в монокультуре грибов обнаружили 29,8% трансформированных клеток. Установлено, что экзометаболиты 65,7% штаммов E. faecalis снижают продукцию каталазы у C. albicans. Исходный уровень каталазы у интактных культур C. albicans в среднем составляет 1,02 мкмоль/мин оптической плотности, после обработки экзометаболитами E. faecalis снижается до 0,55 мкмоль/мин, т.е. на 46,1% (p < 0,05).
Выводы. K. pneumoniae и E. faecalis проявляют антагонизм к C. albicans c разной степенью выраженности. Мишенями для факторов антагонизма факультативной микробиоты у C. albicans являются морфологическая трансформация и продукция каталазы.
420-427
К оценке этиологической значимости бактерий, детектированных в генитальном тракте мужчин
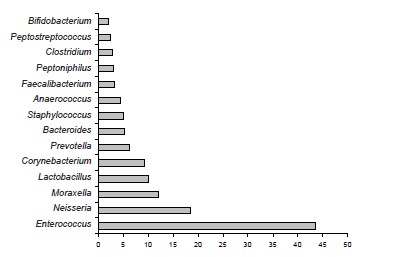
Аннотация
Введение. В настоящее время отмечается возрастающее значение микробных ассоциаций в патогенезе генитальных воспалительных заболеваний. Однако нерешенными остаются вопросы расшифровки таксономической принадлежности и диагностической значимости детектируемых при этом бактерий.
Цель исследования — пересмотр диагностической значимости количественного подхода при определении этиологической роли условно-патогенных микроорганизмов в андрологии.
Материалы и методы. Для исследования использовали пробы эякулята и/или отделяемого из уретры 15 мужчин, состоявших в бесплодном браке, 12 — с подтверждённым диагнозом «острая генитальная гонококковая инфекция». Проводили классическое бактериологическое исследование. Метагеномное исследование 16S рибосомальной РНК образцов осуществлено в отделе коллекционных культур ФБУН ГНЦ ПМБ. Для стандартизации распределения образцов по группам с учётом показателей альфа-разнообразия и концентрации путресцина использовали размах вариации и среднее линейное отклонение.
Результаты. Микробный пейзаж эякулята характеризовался преобладанием представителей родов Enterococcus, Neisseria, Lactobacillus, Corynebacterium, Prevotella, Bacteroides. В эякуляте детектировали устойчивые ассоциации E. faecalis и M. osloensis. При использовании культурального метода представители рода Moraxella не были выделены ни в одной пробе. Показано, что разночтения могут не только затрагивать количественные показатели, но и выявлять несоответствия качественной оценки детектированных генетических маркеров с результатами идентификации в бактериологическом исследовании отдельных представителей соименных и фенотипически сходных таксонов.
Обсуждение. Результаты настоящего исследования указывают на то, что при меньшем разнообразии у условно-патогенных микроорганизмов больше возможностей для реализации своего патогенного потенциала. С другой стороны, в составе сложного сообщества, когда альфа-разнообразие больше, его реализация затруднена из-за сложных межмикробных отношений, с одной стороны, и необходимостью выживать ‒ с другой.
Заключение. Наиболее перспективным следует признать комплексное использование культуромики и метагеномики с учётом сравнительного статистического анализа получаемых качественных и количественных показателей.
428-435
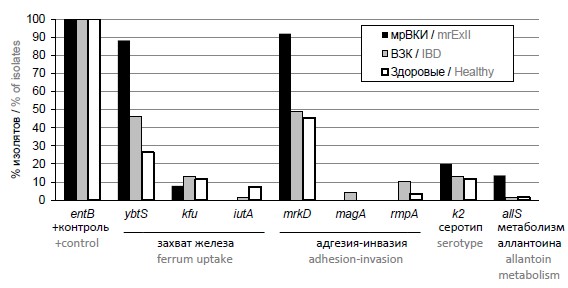
Оценка наличия генов, связанных с вирулентностью, у изолятов Klebsiella pneumoniae, выделенных из микробиоты больных и «практически» здоровых людей, с применением метода мультиплексной полимеразной цепной реакции
Аннотация
Введение. Klebsiella pneumoniae в микробиоте человека может быть представлена как комменсальными, так и высокопатогенными штаммами, например, с гипермукоидным фенотипом. Данный фенотип характеризуется определёнными генетическими детерминантами, позволяющими выявить патогенный потенциал изолятов молекулярно-генетическим методом ПЦР.
Цели и задачи: сравнить патогенный потенциал изолятов K. pneumoniae, полученных от «практически» здоровых лиц, пациентов с воспалительными заболеваниями кишечника (ВЗК), и штаммов, выделенных из биологического материала с внекишечными инфекциями (ВКИ), посредством детекции генов, связанных с вирулентностью.
Материалы и методы. Исследование проводили с применением набора олигонуклеотидов для мультиплексного анализа — 8 генов, предположительно ассоциированных с вирулентностью, с функцией захвата железа (ybtS, kfu, iutA), адгезии и инвазии (mrkD), гипермукоидного фенотипа и вирулентных штаммов (mrkD, magA, rmpA, k2) и метаболизма аллантоина — продукта расщепления пуринов и мочевой кислоты (allS). Анализу подвергли 69 изолятов, выделенных из микробиоты кишечника пациентов с ВЗК, 68 изолятов из кишечной микробиоты «практически» здоровых людей и 25 мультирезистентных изолятов, выделенных из крови, мочи, операционных ран, бронхоальвеолярного лаважа пациентов с ВКИ.
Результаты. Установлено, что 4 из 8 исследованных генов были ассоциированы с различными болезненными состояниями, диагностируемыми у пациентов. Ген сидерофора ybtS достоверно чаще встречался у изолятов, выделенных как у больных с ВЗК (р = 0,024), так и с мультирезистентными ВКИ (p < 0,001). У изолятов ВЗК также достоверно чаще представлен ген гипермукоидного фенотипа rmpA (р = 0,038). У мультирезистентных внекишечных изолятов наиболее достоверно представлены адгезин mrkD (р ≤ 0,001) и allS (р = 0,032).
Заключение. Изоляты K. pneumoniae от пациентов с ВКИ имели наибольший патогенный потенциал, а изоляты из кала «практически» здоровых лиц — наименьший. Наиболее часто встречающиеся гены вирулентности связаны с адгезией и гипермукоидным фенотипом.
436-444
Изучение микробных факторов при обострении полипозного риносинусита
Аннотация
Введение. Полипозный риносинусит (ПРС) считается мультифакторным заболеванием. Имеются данные по участию в инициации и развитии воспалительного процесса грибов и вирусов, о влиянии суперантигенов, биоплёнок и микробиоты на рост полипов в околоносовых пазухах. Обострение заболевания у больных ПРС приводит к значительному снижению качества жизни.
Цель исследования — исследовать бактериальную составляющую микробиоты слизистой оболочки носа и околоносовых пазух у больных с ПРС в период ремиссии и во время обострения.
Материалы и методы. Обследовано 83 человека с ПРС: 44 пациента в период ремиссии, 39 человек в период обострения. У всех пациентов проводилась качественная и количественная оценка бактериальной составляющей микробиоты полости носа.
Результаты. Достоверной разницы в количественном и качественном составе микробиоты полости носа в период обострения и ремиссии воспалительного процесса, а также до и после лечения обострения ПРС нет. Количественная оценка идентифицированных микроорганизмов находилась в подавляющем большинстве случаев в пределах нормы в период обострения и ремиссии воспалительного процесса.
445-452
Нуклеотидные тетрамеры TCGA и CTAG: вирусные ДНК и генетический код (гипотеза)
Аннотация
Введение. Литературные и наши собственные данные показывают, что в частотных профилях (ЧП) герпесвирусных ДНК тетрануклеотиды CTAG и, в меньшей степени, TCGA выделяются среди других полных, билатерально симметричных тетрануклеотидов заметно более низкими значениями концентраций.
Цель работы — сравнительный анализ ЧП тетрануклеотидов CTAG и TCGA в вирусных ДНК.
Материалы и методы. Проанализированы ЧП и другие особенности указанных двух тетрамеров в ДНК не менее одного вида вирусов каждого рода (или субсемейства, если оно не классифицировано по родам) в соответствии с ограничениями по размеру (не ниже 100 000 пар оснований) — всего свыше 200 видов вирусов. Для анализа использованы инструменты GenBank.
Результаты. Описаны две группы формальных особенностей тетрамеров TCGA и CTAG. Одна из них относится к результатам анализа ЧП этих тетрануклеотидов в вирусных ДНК и показывает, что в ДНК с GC:AT > 2 имеют место определённые симметрии ЧП nCGn при частом нарушении таких симметрий в ЧП nTAn из-за недопредставленности CTAG. Другая группа особенностей этих тетрамеров демонстрирует различия их ЧП в полных ДНК вирусов и в их геномах (кодирующей части, которая у некоторых исследованных вирусов достигает 80%, делая анализ их ДНК более убедительным, нежели анализ ДНК клеточных форм жизни) и указывает на возможную роль этих тетрамеров в происхождении универсального генетического кода.
Обсуждение. Предполагается, что генетический код первоначально формировался на основе некоторого преобладания C+G в «до-кодовых» ДНК-полимерах с последующей эволюцией стартовых форм кода до конечной фиксированной структуры, в которой тетрамеры TCGA и CTAG занимают центральное место, отражая исходные этапы этой эволюции. Симметрии ЧП nCGn, характерные для «полной» ДНК герпесвирусов рода Simplex, исчезают в цепи вторых кодонных букв генома этих вирусов, косвенно указывая на отличия их функций от функций других букв и подчёркивая целесообразность представления генетического кода в формате каллиграммы, в которой вторая строка не симметрична.
478-493
Первый случай выявления Listeria monocytogenes сиквенс-типов ST7, ST20, ST425 в сточных водах при обследовании водных объектов Вологодской области
Аннотация
Введение. Listeria monocytogenes относится к числу значимых патогенов человека, вызывает различные формы листериоза, в том числе пищевые инфекции, менингиты, неонатальный сепсис, аборты. Листерии распространены во всех регионах мира.
Целью исследования явилось проведение микробиологического мониторинга Listeria monocytogenes в водных объектах вблизи животноводческих предприятий Вологодского района Вологодской области.
Материалы и методы. Выделение культур бактерий осуществляли титрационным и фильтрационным методами, идентифицировали с применением бактериологического, серологического методов, автоматизированных приборных методов, полногеномного секвенирования и биоинформационного анализа.
Результаты. Из 12 проанализированных образцов водных источников (6 образцов — сточные воды, 4 — речная вода, 2 — ливневые воды) выделены 3 штамма L. monocytogenes и один штамм L. innocua. Полногеномное секвенирование 3 штаммов L. monocytogenes установило их принадлежность к эволюционной линии II, к 3 сиквенс-типам и 2 серогруппам: ST425(1/2a-3a), ST20(1/2a-3a) и ST7(4a-4c). Показана принадлежность штаммов к категории множественно лекарственно-устойчивых, резистентных к 3 функциональным группам антимикробных препаратов (тетрациклинам, макролидам, сульфаниламидам). В геномах штаммов идентифицированы гены антибиотикорезистентности (fosX, pbp-like, lin, norB, sul), острова патогенности LIPI-1,LIPI-2, гены вирулентности inlABCJ, oatA, ami, gtcA, vip, lisK. У 1 штамма выявлен остров стрессоустойчивости SSI-1.
Заключение. Полученные данные свидетельствуют о контаминации водных источников вблизи животноводческих предприятий штаммами L. monocytogenes, обладающими высоким потенциалом патогенности, который может привести к вспышкам листериоза у людей, что указывает на необходимость тщательного мониторинга водных источников на наличие возбудителя листериоза и проведение профилактических и противоэпидемических мероприятий.
453-464
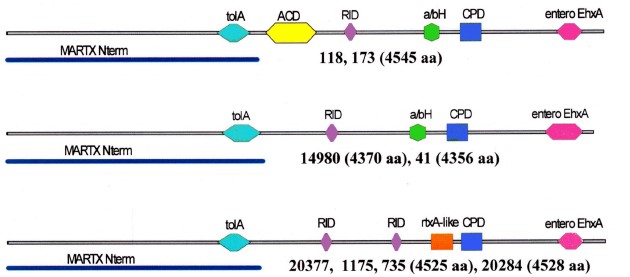
Молекулярно-генетическая характеристика штаммов Vibrio cholerae nonO1/nonO139, выделенных от больных отитами на территории Российской Федерации
Аннотация
Введение. В 2017–2020 гг. в России впервые за много лет от больных отитами были выделены штаммы Vibrio cholerae nonO1/nonO139 (НАГ-вибрионов).
Цель работы — биоинформационный анализ полных геномов (WGSs) и отдельных генов штаммов НАГ-вибрионов — возбудителей отитов, выделенных в России.
Материалы и методы. Анализ WGSs 8 клинических изолятов НАГ-вибрионов, полученных на платформе «MiSeq Illumina», проводили с использованием программ «BioEdit», «BLASTN», «BLASTP», «Vector NTI»; антибиотикоустойчивость определяли согласно МУК 4.2.2495-09.
Результаты. Штаммы различались по содержанию SNP, наборам детерминант факторов патогенности/ персистенции и их аллелям. Все были лишены профагов CTX, preCTX, RS1, острова патогенности VPI, гена термостабильного токсина, мобильных элементов, связанных с антибиотикорезистентностью, острова пандемичности VSP-I; 2 штамма содержали остров VSP-II. В разных сочетаниях выявлены гены ряда протеаз, cholix-токсина, кластер системы секреции 3-го типа (T3SS), дополнительные кластеры T6SS. Продукты изменённых генов сохраняли либо утрачивали характерные активные домены. В цитотоксине MARTX 6 штаммов отсутствовал ключевой домен ACD, у 4 выявлен новый домен rtxA-like. Кластеры генов биоплёнкообразования варьировали по стуктуре. Присутствие генов антибиотикорезистентности не всегда коррелировало с антибиотикограммами. Все штаммы были чувствительны к большинству антибиотиков, но некоторые проявляли резистентность к 1–4 препаратам.
Выводы. Все изученные штаммы — возбудители отитов, несмотря на выявленные различия, обладают достаточными наборами детерминант, ответственных за реализацию патогенетического и персистентного потенциала. В связи с несовпадением генотипических и фенотипических показателей антибиотикорезистентности при выборе препаратов для этиотропной терапии НАГ-инфекций следует полагаться в основном на фенотип. Выявление на территории России больных отитами, вызванными НАГ-вибрионами, указывает на целесообразность включения тестов на их присутствие в схему бактериологического анализа при внекишечных инфекциях и в случаях их выделения — оперативного определения чувствительности к антибиотикам.
465-477
НЕКРОЛОГИ
 494-495
494-495
ЮБИЛЕИ
 496-497
496-497
ХРОНИКА
498-498
Регистрационный номер и дата принятия решения о регистрации СМИ: ПИ № ФС77-75442 от 01.04.2019 г.