


Characterization of the molecular genetic properties of epidemic strains of Klebsiella pneumoniae and Staphylococcus aureus, the pathogens of healthcare-associated infections circulating in the Nizhny Novgorod region
- Authors: Solovyeva I.V.1, Tochilina A.G.1, Belova I.V.1, Zaitseva N.N.1, Kucherenko N.S.2, Sadykova N.A.2, Molodtsova S.B.1, Kropotov V.S.1
-
Affiliations:
- Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
- Department of the Federal Service for Surveillance on Consumer Rights Protection and Human Welfare in the Nizhny Novgorod region
- Issue: Vol 102, No 5 (2025)
- Pages: 560-570
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18975
- DOI: https://doi.org/10.36233/0372-9311-678
- EDN: https://elibrary.ru/LSVTWH
- ID: 18975

Cite item

Abstract
Introduction. Molecular epidemiological monitoring is aimed at obtaining up-to-date information on the genetic variants of healthcare-associated infections (HAIs) circulating in the region. Currently, special attention is being paid to monitoring representatives of the ESKAPE group, as they are a frequent cause of HAIs, complicate the course of the underlying disease, and are becoming an increasingly serious threat to the health and lives of patients due to their complex pathogenicity genes and diverse antibiotic resistance mechanisms.
The aim of the study is to analyze the results of whole-genome sequencing of epidemic strains of pathogens of HAIs — Klebsiella pneumoniae ssp. pneumoniae and Staphylococcus aureus — circulating in the Nizhny Novgorod region.
Materials and methods. Classical bacteriological methods, MALDI-TOF mass spectrometry, whole-genome sequencing, and bioinformatics methods were used.
Results. In-depth analysis revealed the circulation of a population of classical K. pneumoniae strains of sequence type (ST) 3-K type (K) 3 in the neonatal intensive care unit, containing a number of virulence genes and the blaSHV-1 beta-lactamase. Circulation of a population of K. pneumoniae strains of the convergent pathotype ST395 and K39 was detected in a multidisciplinary hospital, and strains of the convergent pathotype K. pneumoniae ST395-K2, K47, as well as strains of the classical pathotype K. pneumoniae ST5209-K35, ST441-K62, ST147-K64, containing a spectrum of pathogenicity genes and beta-lactamases in their genome, including New Delhi metallo-beta-lactamase blaNDM-1, were identified. S. aureus strains associated with catheter-associated bloodstream infections have significant pathogenic potential, belonging to 13 different STs and 19 spa types (t). Circulation of methicillin-resistant (SCCmec IV, ST8, t008) and methicillin-susceptible (ST1, t127) staphylococcal strains has been detected in hemodialysis centers and departments.
Conclusion. The data obtained indicate the circulation of convergent and classical strains of K. pneumoniae and virulent strains of S. aureus in medical and preventive organizations, which justifies the need for molecular epidemiological monitoring.
Full Text
Introduction
Molecular epidemiological monitoring is becoming an integral task in organizing a system for epidemiological surveillance of infectious diseases, as it allows for tracking the circulation of opportunistic microorganisms and timely identification of signs of potential outbreaks, which include: isolation of a homogeneous spectrum of microorganisms from examined individuals; an increase in the incidence of infectious diseases caused by a single species or group of species of pathogens; and an increase in the detection rate of hospital strains. The objectives include monitoring the population structure of infectious agents, including healthcare-associated infections (HAIs), MLST typing of strains, analysis of pathogenicity genes and antibiotic resistance determinants, detection of new variants of hospital strains, and observation of their variability to assess epidemiological forecasting and justify timely intervention in the course of the epidemic process [1].
ESKAPE pathogens are a frequent cause of healthcare-associated infections, complicate the course of the underlying disease, and pose a serious threat to the health and life of patients, as they are able to quickly adapt and find new ways to resist the effect of drugs, disinfectants and antiseptics, and also transmit this ability to other pathogens at a genetic level [2, 3].
Among the bacteria that cause hospital infections, Klebsiella pneumoniae and Staphylococcus aureus are the leading causes, with an increasing proportion of carbapenem-resistant K. pneumoniae and the widespread prevalence of methicillin-resistant Staphylococcus aureus (MRSA) belonging to the MRSA group, which can cause outbreaks and lead to catheter-associated bloodstream infections (CABSI) [4, 5].
The aim of the study is to analyze the results of whole-genome sequencing of epidemic strains of pathogens of healthcare-associated infections — K. pneumoniae ssp. pneumoniae and S. aureus, circulating in the Nizhny Novgorod region.
Materials and methods
Strains under study
55 epidemic strains of pathogens were studied: 17 strains of K. pneumoniae ssp. pneumoniae and 38 strains of S. aureus. Based on their place of isolation, the strains were divided into three groups:
The 1st group consisted of 7 K. pneumoniae strains isolated from sick children (gastric contents) (n = 4) in the neonatal intensive care unit of a pediatric hospital, and from equipment and care items (swabs from suction tubing, feeding syringe) (n = 3);
The 2nd group – 10 strains of K. pneumoniae isolated from patients in the multidisciplinary hospital departments (wound discharge) (n = 9) and from the department's external environment (swab from the intensive care unit sink faucet) (n = 1);
The 3rd group – 38 strains of S. aureus, including 31 strains from patients with CABSI who were receiving outpatient treatment at hemodialysis centers in the city and region and were hospitalized in medical organizations in the city (blood, wound at the catheter site, peritoneal fluid, nasal swab); 3 strains isolated from medical personnel (nasal swab), and 4 from the medical organization environment (equipment swabs).
Cultivation and identification of bacteria
Isolation of strains of conditionally pathogenic microorganisms was carried out using the classical bacteriological method, and identification was performed by MALDI-TOF mass spectrometry using an Autoflex mass spectrometer (Bruker Daltonics). The susceptibility of bacteria to antibiotics was studied using the disk diffusion method on "Nutrient Medium for Determining the Susceptibility of Microorganisms to Antibacterial Drugs — Mueller-Hinton II Agar (State Research Center for Applied Microbiology and Biotechnology of Rospotrebnadzor) using extended sets of disks for enterobacteria (set No. 7) and staphylococci (set No. 14) (Pasteur Research Institute of Epidemiology and Microbiology). The susceptibility of the strains to ceftazidime-avibactam was studied using ceftazidime + avibactam 10/4 mcg disks (Mast Group), and to tigecycline using tigecycline 15 mcg disks (Mast Group). The assessment was conducted in accordance with the clinical guidelines "Determination of the Susceptibility of Microorganisms to Antimicrobial Drugs"1.
Whole-genome sequencing
Libraries were prepared using the TrueSeq kit (Illumina Inc.), and sequencing was performed on the MiSeq platform (Illumina Inc.). The raw reads were processed using the Trimmomatic utility, while the SPAdes v. 3.11.1 and Prokka v. 1.12 programs were used for de novo read assembly [6, 7]. All nucleotide sequences were deposited in the international GenBank database.
Whole-genome sequence analysis was performed using VFDB2 [8], ResFinder3 [9], the BIGSdb-Pasteur web platform4 [10], the PubMLST resource5 [11], and the Spa-typer and SCCmecFinder programs [12, 13]. Dendrograms were constructed using the maximum likelihood method to determine the genetic distance between microbial strains using the parsnp v. 1.7.4 program. The FastTree 2.1.1 algorithm and the Shimodaira–Hasegawa test [14] were used to assess the primary tree topology. Sequences from the GenBank database were used as references: GCF_000240185.1, GCA_000013425.1, and then the reference genome branch was removed. Phylogenetic trees were visualized using the iTol service [15].
Results
As a result of studying the molecular genetic properties of K. pneumoniae strains of group 1, it was found that all strains possess colibactin gene clusters, yersiniabactin genes, and genes responsible for the formation of type 3 fimbriae, and the rmpA gene, which regulates the hypermucoviscous phenotype, was detected in 3 strains (Table). A species-specific antibiotic resistance (ABR) determinant was identified in the genomes of all strains – the beta-lactamase blaSHV-1, which confers natural resistance of microorganisms to aminopenicillins – ampicillin and amoxicillin. Phenotypically, the strains were susceptible to all antibiotics from other groups: 3rd–5th generation cephalosporins (cefazolin, cefotaxime, ceftriaxone, ceftazidime, ceftazidime/avibactam, cefepime, ceftaroline), aminoglycosides (gentamicin), fluoroquinolones (ciprofloxacin), carbapenems (ertapenem, imipenem, meropenem), monobactams (aztreonam), polymyxins (colistin), tetracyclines (tigecycline), and trimethoprim/sulfamethoxazole. As a result of the analysis of constitutional genes (housekeeping genes) and wzi gene alleles, the strains were identified as belonging to ST 3 and capsular type K 3 (Table).
Molecular genetic characterization of K. pneumoniae strains isolated in the neonatal intensive care unit
Strain | Sequence type (ST) | Capsule type (К) | Pathogenicity genes | ABR genes | |||
hypermucoid phenotype regulator genes | colibactin synthesis gene | Yersiniabactin synthesis genes | type 3 fimbriae genes | ||||
n = 3 K. pn 849 JAVGJO000000000 K. pn 852 JAVHUD000000000 K. pn 862 JAVBWS000000000 | 3 | 3 | rmpA | clbABCDEFGHLMNOPQ | fyuA, irp1,2, ybtAEQPSTUX | mrkABCDFHIJ | blaSHV-1 |
n = 4 K. pn 850 JAVCZJ000000000 K. pn 854 JAVGJN000000000 K. pn 863 JAVBWT000000000 K. pn 893 JAVHUE000000000 | 3 | 3 | – | clbABCDEFGHLMNOPQ, | fyuA, irp1,2, ybtAEQPSTUX | mrkABCDFHIJ | blaSHV-1 |
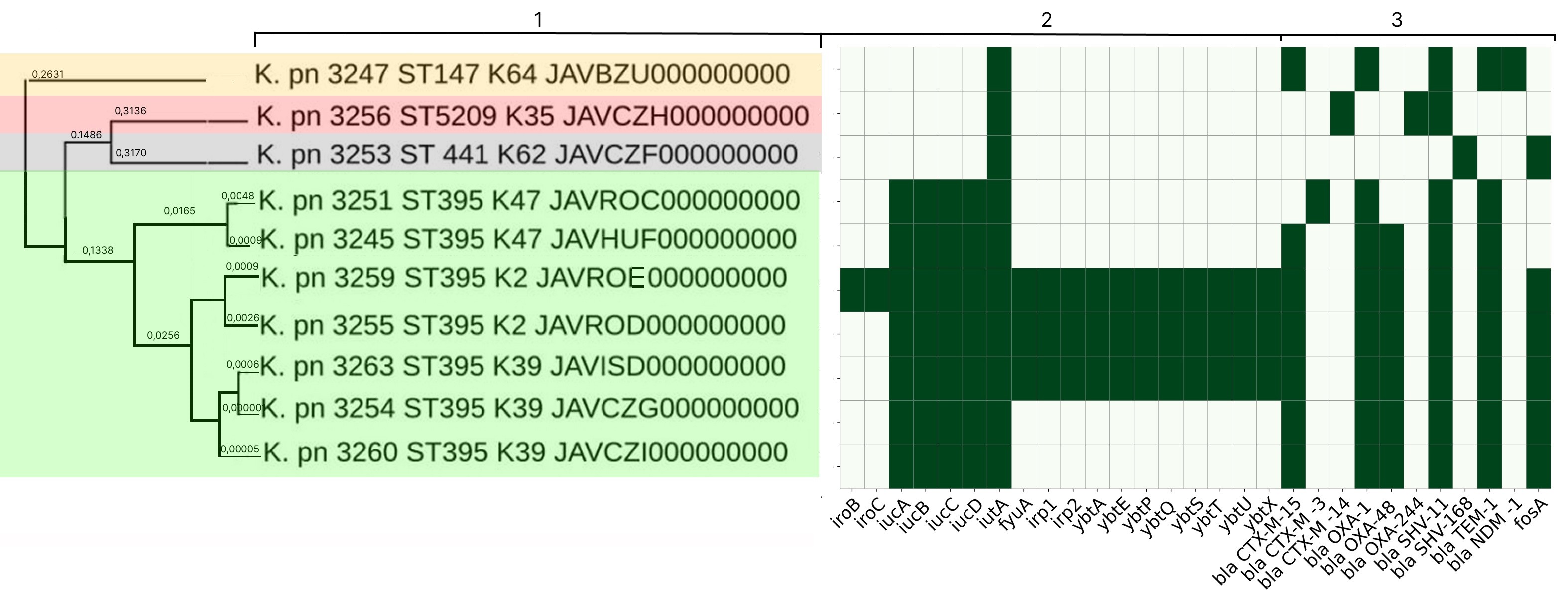
K. pneumoniae strains of group 2 differed in their molecular genetic properties. Complete aerobactin siderophore gene clusters (iucABCD and iutA) were found in the genomes of K. pneumoniae strains 3254, 3260 and 3263, while K. pneumoniae strain 3263 was distinguished by the presence of the yersiniabactin gene cluster (fyuA, irp1,2, ybtAEPQSTUX) (Fig. 1). All strains were found to have a spectrum of ARGs – beta-lactamases and carbapenemases blaTEM-1, blaCTX-M-15, blaSHV-11, blaOXA-1, blaOXA-48, and the fosfomycin resistance determinant – fosA. This explains the fact that all 3 strains had an MDR phenotype and were resistant to penicillins, cephalosporins, aminoglycosides, fluoroquinolones, monobactams and carbapenems (ertapenem). Based on the analysis of constitutional genes and wzi gene alleles, the strains were identified as belonging to ST395 and K39.

Fig. 1. Phylogenetic tree for genomes of K. pneumoniae strains of group 2.
1 — microorganism type, strain number, ST, K-type, GenBank database accession number; 2 — determinants of pathogenicity genes (green — presence of the trait, white — absence); 3 — determinants of ABR (green — presence of the trait, white — absence)
The virulence gene spectrum of K. pneumoniae strains 3245 and 3251, like that of K. pneumoniae strains 3254 and 3260, was represented by the aerobactin gene cluster (iucABCD and iutA) (Fig. 1). K. pneumoniae strain 3245 was found to have an identical spectrum of antibiotic resistance genes (blaTEM-1, blaCTX-M-15, blaSHV-11, blaOXA-1, blaOXA-48) in its genome, with the exception of fosA. This strain had the same antimicrobial susceptibility and resistance phenotypes as the strains described above. K. pneumoniae 3251 was characterized by the presence of the blaCTX-M-3 determinant, the absence of the blaOXA-48 and blaCTX-M-15 genes, and was susceptible to carbapenems. K. pneumoniae 3245 and 3251 also belonged to ST395, but differed from K. pneumoniae strains 3254, 3260, and 3263 in their wzi gene alleles and belonged to K47.
K. pneumoniae strains 3255 and 3259 were characterized by the presence of complete aerobactin (iucABCD and iutA) and yersiniabactin (fyuA, irp1,2, ybtAEPQSTUX) gene clusters, with the salmochelin gene (iro) being detected in K. pneumoniae 3259. Their ABR gene spectrum was identical to the spectrum of determinants in K. pneumoniae 3254, 3260, 3263, 3245 (blaTEM-1, blaCTX-M-15, blaSHV-11, blaOXA-1, blaOXA-48, fosA). K. pneumoniae 3255 and 3259 also had an MDR phenotype and were resistant to penicillins, cephalosporins, aminoglycosides, fluoroquinolones, carbapenems (ertapenem), and tigecycline. These strains also belonged to ST395, but to K2.
Only one aerobactin cluster determinant, iutA, was found in K. pneumoniae strains 3247, 3253, and 3256 (Fig. 1). K. pneumoniae isolate 3256 contained the beta-lactamases blaCTX-M-14, blaOXA-244 and blaSHV-11 in its genome, exhibited phenotypic resistance to cephalosporins and aminoglycosides, and was susceptible to carbapenems. Its affiliation with ST5209 and K35 was established. K. pneumoniae 3253 was found to have 2 AMR determinants: blaSHV-168 and fosA. The strain was susceptible to all groups of antibacterial drugs except aminopenicillins and was classified as ST441 and K62. In the genome of K. pneumoniae 3247, the blaNDM-1, blaTEM-1, blaCTX-M-15, blaSHV-11 and blaOXA-1 genes were detected. The strain was phenotypically resistant to penicillins, cephalosporins, aminoglycosides, fluoroquinolones, all carbapenem group drugs (ertapenem, imipenem, meropenem), and susceptible to colistin, tigecycline, and ceftazidime-avibactam, and was classified as ST147 and K64.
Adhesin genes, a type VII secretion system, and gamma-hemolysins were identified in the genomes of all group 3 S. aureus strains.
Strains S. aureus 2226, 3092, 3110, 2211, 3196, 3197, and 3198 were characterized by an identical spectrum of virulence genes, which included, in addition to those listed above, the serine protease genes splABCDE, the immune evasion genes sak, scn, enterotoxin A sea, exfoliative toxin eta, and leukotoxins lukDE. SCCmec type IV cassettes were detected in strains 3196, 3197, 3110 and 3092, the blaZ beta-lactamase gene was identified in S. aureus strains 3198, 2211 and 2226, and the erythromycin and chloramphenicol resistance genes ermC and cat were found (Fig. 2). The strains showed phenotypic resistance to oxacillin and cefoxitin, which may indicate their belonging to the MRSA group. As a result of MLST typing and analysis of the protein A gene sequence repeats, their belonging to ST8 and spa-type (t) t008 was established. Also belonging to ST8, but to t024, was the S. aureus strain 3107, which was distinguished by the absence of the SCCmec cassette.

Fig. 2. Phylogenetic tree for genomes of S. aureus strains of group 3.
1 — microorganism type, strain number, GenBank database accession number, ST, spa type; 2 — pathogenicity gene determinants (green — presence of the trait, white — absence); 3 — antibiotic resistance determinants (green — presence of the trait, white — absence).
Pathogenicity genes splABCDE, eta and lukDE were identified in the genomes of S. aureus strains 3086, 3087 and 3088, while S. aureus 2203 and 2213 were characterized by the presence of sak and scn genes (Fig. 2). The blaZ and ermC antibiotic resistance genes were found in S. aureus 3086, 3087, and 3088, while only blaZ was found in S. aureus 2213. All 5 isolates were resistant to amoxicillin, tetracycline and lincosamides. It was established that strains 3086, 3087, and 3088 belonged to ST1 t127, while S. aureus 2203 and 2213 belonged to ST1 t177.
As a result of analyzing the molecular genetic properties of S. aureus strains 3082, 3094, 3111, 3102, 2212, and 2204, the genes for exfoliative toxin eta and staphylokinase sak were identified, and the absence of serine protease determinants (spl) and leukotoxins D and E (lukDE) was established. Enterotoxin genes sec and sell were found in S. aureus 3082; sec, sell and selo were found in 3094. The blaZ gene was present in S. aureus 3082, 3111, 3094, and 3082; the blaZ gene and the aminoglycoside resistance determinant aph(3')-III were present in strain 3094; and aph(3')-Ia was present in S. aureus 2212. All 5 isolates were phenotypically resistant to amoxicillin and amikacin. The strains were identified as belonging to ST45, but to different spa-types: t102, t362, t8416, t5599, t5132 (Fig. 2).
S. aureus strains 3084, 3085, and 2219 were characterized by the presence of the pathogenicity genes splABCDE, eta, lukDE and blaZ. Genes for enterotoxins sei, sej, selo and selr were detected in 3085 strains. All strains were phenotypically resistant to amoxicillin and were identified as belonging to ST5, t002, and t688 (Figure 2). S. aureus strains 3096 and 3089 were characterized by the presence of the splABCDE, lukDE, sak, scn, and blaZ genes; strain 3096 contained the tsst gene, while strain 3089 contained the sea gene. They are also resistant to amoxicillin and were classified as ST707 and ST6, although the program used in the study did not allow their spa-type to be determined. S. aureus 3100 and 3133 were characterized by the absence of serine protease genes (spl) and leukotoxins D and E (lukDE); strain 3133 was resistant to oxacillin and cefoxitin, and a type IV SCCmec cassette was found in its genome. Both strains belonged to ST398, t571 and t011 (Fig. 2).
The remaining 12 strains belonged to different ST and spa-types. The genes splABCDE, eta, lukDE and sak were detected in S. aureus strains 2210, 3104, 3102 and 2207, and the toxic shock syndrome gene tsst was detected in S. aureus 3104. The strains belonged to ST97 and different spa types: t267, t3380, t11521 (Fig. 2). S. aureus strains 2208, 2209 and 2214 were characterized by the presence of the splABCDE, lukDE, sak, and scn genes and were assigned to ST12 and t156. Genes splABCDE and lukDE were identified in S. aureus 2206, 2217 and 2215, and their belonging to ST49 and ST1027 was established, but the program used in the study did not allow for the determination of their spa-types. Strain S. aureus 2222 was found to have the splABCDE, lukE, sak, scn, eta and tsst genes and was classified as ST426 and t764. S. aureus 3295 was characterized by the presence of sak and eta determinants and was classified as ST 4. All 12 strains lacked antibiotic resistance determinants and exhibited phenotypic susceptibility to antibacterial drugs from all groups.
In total, strains of S. aureus belonging to 13 different STs and 19 spa types were isolated from patients with CABSI, medical personnel and the external environment of the city and region.
Discussion
Circulation of the pathogen refers to its continuous and sequential transmission from one susceptible organism to another, ensuring its existence as a biological species, as well as the spread of the pathogen within healthcare facilities, characterized by the colonization of environmental surfaces and the involvement of patients and staff.
One of the most important criteria for a hospital strain is its belonging to a homogeneous (uniform in composition) population of circulating microorganisms [16]. The homogeneity of a population can be most reliably assessed by studying the genetic characteristics of the strains, which involves identifying and analyzing virulence genes, antibiotic resistance genes, and determining the ST through the analysis of alleles of housekeeping genes. For the genetic typing of K. pneumoniae, determining their K-type, which depends on the sequence of the wzi gene encoding a surface protein involved in capsule assembly on the cell's outer membrane, is of significant importance. Establishing the belonging of strains to one of the known pathotypes is also important [17].
In recent years, the existence of three K. pneumoniae pathotypes has been recognized: hypervirulent (hvKp), classical (cKp), and convergent (hv-MDRKp). The hypervirulent pathotype is associated with the development of serious invasive infections in healthy immunocompetent individuals. Currently, the main characteristic correlated with hypervirulence is the secretion of the siderophores aerobactin, salmochelin, yersiniabactin and the exotoxin colibactin [18]. The classical pathotype is globally widespread; these Klebsiella are representatives of the human microbiome, cause diseases in weakened patients, and are among the leading causes of nosocomial infections. Their genome invariably contains a complex of beta-lactamases and carbapenemases, and individual pathogenicity genes (excluding aerobactin and salmochelin) can also be detected. K. pneumoniae of the convergent pathotype combine the characteristics of hypervirulent and classical Klebsiella, meaning they have high pathogenic potential and multiple antibiotic resistance, and are capable of causing disease in both healthy immunocompetent individuals and immunocompromised patients [17, 18].
K. pneumoniae strains of group 1 isolated from sick children in the neonatal intensive care unit, as well as from equipment and care items, were similar in their virulence determinants and did not contain the aerobactin and salmochelin genes. However, the rmpA gene (hypermucoid phenotype) was detected in 3 strains, which was previously associated with hypervirulence. Currently, it is recognized that it is advisable to assess the entire complex of pathogenicity genes, and the presence of this determinant is not a significant indicator [5, 18]. The presence of the gene in only 3 out of the 7 strains studied (table) can be explained by its plasmid origin and high mobility within the microorganism population [19]. One beta-lactamase, blaSHV-1, was identified in the genomes of all strains. Molecular typing revealed that the strain population belonged to a rare sequence type, ST3, and capsule type K3, which had not been previously isolated in Russia (only 16 isolates of this ST are registered in the BIGSdb-Pasteur database).
The homogeneity of the molecular-genetic and phenotypic properties of the strains isolated from patients and the external environment indicates the circulation of a population of classical K. pneumoniae strains within the hospital. The identification of this population is unfavorable from an epidemiological perspective and confirms the fact that cases of HAIs in the neonatal intensive care unit can be associated not only with hypervirulent strains of K. pneumoniae but also with classical strains that do not possess multiple antibiotic resistance and a wide range of pathogenicity genes [20]. This necessitates continuous molecular-epidemiological monitoring of K. pneumoniae strains belonging to different pathotypes in the microbiota of newborns, mothers, medical personnel, and surrounding objects in neonatal units of pediatric hospitals and perinatal centers, studying their properties, as well as integrating the data into the VGARus Russian database.
Analysis of K. pneumoniae strains from group 2 isolated in a multidisciplinary hospital revealed that strains K. pneumoniae 3254, 3260, and 3263 had an identical pathogenicity and antibiotic resistance gene spectrum, as well as the same antibiotic resistance phenotype. These strains belonged to ST395-K39 and were classified as a convergent pathotype due to the presence of aerobactin and a spectrum of beta-lactamase and carbapenemase genes. The strains were isolated from both patients and the external environment, indicating the circulation of K. pneumoniae ST395-K39 strains within the hospital. The presence of the yersiniabactin gene cluster in K. pneumoniae 3263 can be explained by horizontal gene transfer processes associated with a transposon, which has high mobility [21]. K. pneumoniae strains 3255 and 3259 (ST395-K2) and K. pneumoniae 3245 and 3251 (ST395-K47) were isolated in single cases only from patients, contained the aerobactin gene and AMR determinants in their genomes, and were also classified as a convergent pathotype.
Thus, 7 out of 10 strains isolated in a multidisciplinary hospital belonged to ST395, which could be considered evidence of their circulation. This is consistent with scientific literature data on the widespread distribution of this ST, among which strains with significant pathogenic potential are frequently found, capable of causing severe systemic infections [5, 17]. However, in-depth analysis of the strains' molecular genetic properties revealed their heterogeneity even within a single ST and led to the conclusion that only the K. pneumoniae ST395-K39 strain populations were circulating in the hospital.
All other K. pneumoniae strains: 3256 (ST5209-K35), 3253 (ST 441-K62), 3247 (ST147-K64) were classified as classical and were isolated only in isolated cases from patients. It should be noted that the blaNDM-1 gene for New Delhi metallo-beta-lactamase was found in the genome of the K. pneumoniae 3247 strain, which explains this strain's high degree of antibiotic resistance. It is known that this determinant is associated with plasmids and is capable of active horizontal transfer [17], so the detection of such a strain is unfavorable from an epidemiological perspective, as it can lead to the rapid global spread of a polyresistant population within medical facilities.
Within the phylogenetic tree, all strains from group 2 clustered according to their ST and K-types. K. pneumoniae ST 395 strains formed 3 subclusters according to their K-types, and 3 K. pneumoniae ST395-K39 strains were included in a single cluster, uniting strains isolated from patients and the external environment of the medical organization (Fig. 1).
Analysis of pathogenicity gene spectra, antibiotic resistance profiles, MRSA or MSSA (methicillin-susceptible staphylococci) group affiliation, and ST and spa-typing [22] is crucial for studying the circulation of S. aureus strains. This is based on the analysis of the sequence of repeats in the gene for staphylococcal surface protein A (protein A) [4]. During investigations of local outbreaks of illness, determining the spa-type is an important step, as it allows for differentiation between strains belonging to the same ST.
Molecular genetic analysis of S. aureus group III strains associated with CABSI revealed that the genomes of 7 strains of S. aureus 2226, 3092, 3110, 2211, 3196, 3197 and 3198 contained a type IV SCCmec cassette, they had identical virulence gene profiles, the same resistance phenotypes, and belonged to ST 8 t008.
The strains were isolated from both a healthcare worker and patients diagnosed with CABSI who were receiving outpatient treatment at a hemodialysis center and inpatient treatment at medical institutions in the city, which confirms the fact of their circulation. The difference in strains based on the spectrum of blaZ, ermC and cat determinants (Fig. 2) can be explained by the fact that these genes are located on plasmids and have high mobility [23–25]. According to scientific literature, S. aureus ST8 t008 SCCmec IV strains are common, often associated with HAIs, and have been identified in Russia since the 1990s [4].
Strains S. aureus 3086, 3087 and 3088 are also identical in their pathogenicity genes, determinants, and antibiotic resistance phenotype. They belonged to the MSSA group, were typed as S. aureus ST1 t127, and were isolated from 3 patients (peritoneal catheter exit site). At the same time, strains of S. aureus of other sequence types were isolated from the peritoneal fluid of these same patients — ST5 (t688), ST97 (t267), and ST45 (t8416). This indicates that the population of S. aureus ST1 t127 strains is circulating in this medical organization.
All other group III S. aureus isolates were heterogeneous in their determinant spectrum, antibiotic resistance phenotype, sequence types, and spa types, which prevents an assessment of their epidemiological significance in this study.
Phylogenetic analysis of whole-genome sequences of the strains revealed the presence of five clusters, grouping strains belonging to the same sequence types and clonal complexes (CCs), which are groups of genetically closely related sequence types. S. aureus strains ST8, 97, 12, and 1 formed independent groups (A, B, C, D), while strains of various sequence types: ST6 and ST5 belonging to clonal complex 5 (CC5), S. aureus ST4 and ST45 belonging to CC45, as well as strains ST49, ST1027, ST707, ST398 and ST426 not belonging to specific clonal complexes but having phylogenetic relatedness to each other, were included in a single large cluster E (Fig. 2).
Thus, in the hemodialysis units of the city and region, 13 different sequence types and 19 spa-types of S. aureus strains were identified, and the circulation of populations of epidemic S. aureus MRSA (SCCmec IV) of molecular type ST8 t008 and S. aureus MSSA of molecular type ST1 t127 was demonstrated.
Conclusion
As a result of the study conducted in the Nizhny Novgorod region, a large genetic diversity of K. pneumoniae and S. aureus strains was identified. In-depth analysis revealed the circulation of a population of classical K. pneumoniae ST3-K3 strains in the neonatal intensive care unit of a pediatric hospital, isolated from the gastrointestinal tracts of children, from medical equipment and products, containing a number of virulence genes and the blaSHV-1 beta-lactamase.
The circulation of strains of the convergent K. pneumoniae ST395-K39 pathotype was detected in a multidisciplinary hospital, and strains of K. pneumoniae ST395-K2, ST395-K47, ST5209-K35, ST441-K62, ST147-K64 were identified, containing a spectrum of pathogenicity genes and beta-lactamases in their genome, including the New Delhi metallo-beta-lactamase blaNDM-1, which could lead to the formation and spread of a polyresistant clone in the hospital, capable of replacing the circulating pathogen and causing outbreaks of healthcare-associated infections.
Circulation of S. aureus MRSA (SCCmec IV) ST8 t008 and S. aureus MSSA ST1 t127 populations was identified in the hemodialysis units of the city and region, and other S. aureus strains belonging to 11 different STs- and 17 spa-types, potentially capable of forming hospital clones and spreading widely within the medical facility, were also detected. In this regard, to prevent the occurrence and spread of healthcare-associated infections (HAIs), it is necessary to conduct mandatory continuous microbiological monitoring in hospitals, an integral part of which should be molecular epidemiological monitoring aimed at obtaining up-to-date information on the genetic variants of circulating pathogens, including K. pneumoniae and S. aureus.
1 MACMACH Recommendations version 2024. URL: https://www.antibiotic.ru/minzdrav/category/clinical-recommendations
2 Virulence factor database. URL: http://www.mgc.ac.cn/VFs
3 ResFinder. URL: http://genepi.food.dtu.dk/resfinder
4 Institut Pasteur. Klebsiella pneumoniae species complex.
URL: https://bigsdb.pasteur.fr/klebsiella
5 PubMLST. MLST Database. Staphylococcus aureus.
URL: https://pubmlst.org/organisms/staphylococcus-aureus
About the authors
Irina V. Solovyeva
Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Email: lab-lb@yandex.ru
ORCID iD: 0000-0002-3136-9500
D. Sci. (Biol.), leading researcher, Head, Laboratory of the human microbiome and means of its correction
Russian Federation, Nizhny NovgorodAnna G. Tochilina
Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Author for correspondence.
Email: lab-lb@yandex.ru
ORCID iD: 0000-0001-7753-5730
Cand. Sci. (Biol.), senior researcher, Laboratory of the human microbiome and means of its correction
Russian Federation, Nizhny NovgorodIrina V. Belova
Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Email: lab-lb@yandex.ru
ORCID iD: 0000-0003-3402-1160
Cand. Sci. (Med.), leading researcher, Laboratory of the human microbiome and means of its correction
Russian Federation, Nizhny NovgorodNatalya N. Zaitseva
Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Email: nniiem@yandex.ru
ORCID iD: 0000-0001-5370-4026
D. Sci. (Med.), Director
Russian Federation, Nizhny NovgorodNatalia S. Kucherenko
Department of the Federal Service for Surveillance on Consumer Rights Protection and Human Welfare in the Nizhny Novgorod region
Email: sanepid@sinn.ru
ORCID iD: 0000-0002-0509-3459
Head
Russian Federation, Nizhny NovgorodNatalia A. Sadykova
Department of the Federal Service for Surveillance on Consumer Rights Protection and Human Welfare in the Nizhny Novgorod region
Email: sanepid@sinn.ru
ORCID iD: 0000-0001-9412-8678
Deputy Head
Russian Federation, Nizhny NovgorodSvetlana B. Molodtsova
Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Email: lab-lb@yandex.ru
ORCID iD: 0000-0002-4750-5925
researcher, Laboratory of the human microbiome and means of its correction
Russian Federation, Nizhny NovgorodVasiliy S. Kropotov
Academician I.N. Blokhina Nizhny Novgorod Scientific Research Institute of Epidemiology and Microbiology
Email: lab-lb@yandex.ru
ORCID iD: 0000-0002-6903-962X
Cand. Sci. (Biol.), senior researcher, Laboratory of the human microbiome and means of its correction
Russian Federation, Nizhny NovgorodReferences
- Зубков В.В., Любасовская Л.А., Рюмина И.И. и др. Микробиологический мониторинг в системе инфекционного контроля неонатальных стационаров. Российский вестник перинатологии и педиатрии. 2014;59(1):51–6. Zubkov V.V., Lyubasovskaya L.A., Ryumina I.I., et al. Microbiological monitoring of the infection control system of neonatal hospitals. Russain Bulletin of Perinatology and Pediatrics. 2014;59(1):51–6. EDN: https://elibrary.ru/rwimxh
- Михайловская В.С., Селиванова П.А., Кузнецова М.В. Распространённость генов qacEΔ1, qacE, oqxA, oqxB, acrA, cepA и zitB среди мультирезистентных Klebsiella pneumoniae, выделенных в кардиохирургическом стационаре. Журнал микробиологии, эпидемиологии и иммунобиологии. 2024;101(4):502–11. Mihailovskaya V.S., Selivanova P.A., Kuznetsova M.V. Prevalence of qacEΔ1, qacE, oqxA, oqxB, acrA, cepA and zitB genes among multidrug-resistant Klebsiella pneumoniae isolated in a cardiac hospital. Journal of Microbiology, Epidemiology and Immunobiology. 2024;101(4): 502–11. DOI: https://doi.org/10.36233/0372-9311-548 EDN: https://elibrary.ru/fcoyee
- Новикова И.Е., Садеева З.З., Алябьева Н.М. и др. Антибиотикорезистентность и вирулентность карбапенем-устойчивых штаммов Klebsiella pneumoniae, выделенных у детей в реанимационных и хирургических отделениях. Журнал микробиологии, эпидемиологии и иммунобиологии. 2023;100(4):321–32. Novikova I.E., Sadeeva Z.Z., Alyabyeva N.M., et al. Antimicrobial resistance and virulence of carbapenem-resistant Klebsiella pneumoniae strains isolated from children in intensive care and surgical units. Journal of Microbiology, Epidemiology and Immunobiology. 2023;100(4):321–32. DOI: https://doi.org/10.36233/0372-9311-373 EDN: https://elibrary.ru/rmjxsl
- Романов А.В., Дехнич А.В., Эйдельштейн М.В. Молекулярная эпидемиология штаммов Staphylococcus aureus в детских стационарах России. Клиническая микробиология и антимикробная химиотерапия. 2012;14(3):201–8. Romanov А.V., Dekhnich А.V., Edelstein М.V. Molecular epidemiology of Staphylococcus aureus in Russian pediatric hospitals. Clinical Microbiology and Antimicrobial Chemotherapy. 2012;14(3):201–8. EDN: https://elibrary.ru/pcnnlp
- Воронина О.Л., Кунда М.С., Рыжова Н.Н. и др. Геномные особенности резистентных изолятов Klebsiella pneumoniae, выделенных из кровяного русла и ликвора пациентов детского стационара. Журнал микробиологии, эпидемиологии и иммунобиологии. 2023;100(6):399–409. Voronina O.L., Kunda M.S., Ryzhova N.N., et al. Genomic features of resistans Klebsiella pneumoniae, isolated from the bloodstream and cerebrospinal fluid of pediatric hospital patients. Journal of Microbiology, Epidemiology and Immunobiology. 2023;100(6):399–409. DOI: https://doi.org/10.36233/0372-9311-430 EDN: https://elibrary.ru/ylxbdz
- Bankevich A., Nurk S., Antipov D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19(5):455–77. DOI: https://doi.org/10.1089/cmb.2012.0021
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9. DOI: https://doi.org/10.1093/bioinformatics/btu153
- Chen L.H., Yang J., Yu J., et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(1):325–8. DOI: https://doi.org/10.1093/nar/gki008
- Bortolaia V., Kaas R.S., Ruppe E., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020;75(12):3491–500. DOI: https://doi.org/10.1093/jac/dkaa345
- Jolley K.A., Bray J.E., Maiden M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124. DOI: https://doi.org/10.12688/wellcomeopenres.14826.1
- Guo C., Yang X., Wu Y., et al. MLST-based inference of genetic diversity and population structure of clinical Klebsiella pneumoniae, China. Sci. Rep. 2015;5:7612. DOI: https://doi.org/10.1038/srep07612
- Bartels M.D., Petersen A., Worning P., et al. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 2014;52(12):4305–8. DOI: https://doi.org/10.1128/jcm.01979-14
- International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC). Classification of staphylococcal cassette chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob. Agents Chemother. 2009;53(12):4961–7. DOI: https://doi.org/10.1128/aac.00579-09
- Treangen T.J., Ondov B.D., Koren S., et al. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15(11):524. DOI: https://doi.org/10.1186/s13059-014-0524-x
- Letunic I., Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):293–6. DOI: https://doi.org/10.1093/nar/gkab301
- Брико Н.И., Брусина Е.Б., Зуева Л.П. и др. Госпитальный штамм – непознанная реальность. Эпидемиология и вакцинопрофилактика. 2013;(1):30–5. Briko N.I., Brusina E.B., Zueva L.P., et al. Hospital strain — a mysterious reality. Epidemiology and Vaccinal Prevention. 2013;(1):30–5. EDN: https://elibrary.ru/pvsumn
- Агеевец В.А., Агеевец И.В., Сидоренко С.В. Конвергенция множественной резистентности и гипервирулентности у Klebsiella pneumoniae. Инфекция и иммунитет. 2022;12(3):450–60. Ageevets V.A., Ageevets I.V., Sidorenko S.V. Convergence of multiple resistance and hypervirulence in Klebsiella pneumoniae. Russian Journal of Infection and Immunity. 2022;12(3):450–60. DOI: https://doi.org/10.15789/2220-7619-COM-1825 EDN: https://elibrary.ru/ucpmnf
- Комисарова Е.В., Воложанцев Н.В. Гипервирулентная Klebsiella pneumoniae — новая инфекционная угроза. Инфекционные болезни. 2019;17(3):81–9. Komisarova E.V., Volozhantsev N.V. Hypervirulent Klebsiella pneumoniae: a new infectious threat. Infectious diseases. 2019;17(3):81–9. DOI: https://doi.org/10.20953/1729-9225-2019-3-81-89 EDN: https://elibrary.ru/idonjy
- Ali M.R., Yang Y., Dai Y., et al. Prevalence of multidrug-resistant hypervirulent Klebsiella pneumoniae without defined hypervirulent biomarkers in Anhui, China: a new dimension of hypervirulence. Front. Microbiol. 2023;14:1247091. DOI: https://doi.org/10.3389/fmicb.2023.1247091.8
- Устюжанин А.В., Маханёк А.А., Чистякова Г.Н. и др. Сравнительный геномный анализ клинических изолятов Klebsiella pneumoniae, выделенных от новорождённых детей с различными исходами инфекционного процесса в неонатальном периоде. Журнал микробиологии, эпидемиологии и иммунобиологии. 2025;102(1):62–71. Ustyuzhanin A.V., Makhanyok A.A., Chistyakova G.N. et al. Comparative genomic analysis of clinical isolates of Klebsiella pneumoniae isolated from newborns with different outcomes of the infectious process in the neonatal period. Journal of Microbiology, Epidemiology and Immunobiology. 2025;102(1):62–71. DOI: https://doi.org/10.36233/0372-9311-544 EDN: https://elibrary.ru/zxmnbq
- Самойлова А.А., Краева Л.А., Михайлов Н.В. и др. Геномный анализ вирулентности и антибиотикорезистентности штаммов Klebsiella pneumoniae. Инфекция и иммунитет. 2024;14(2):339–50. Samoylova A.A., Kraeva L.A., Mikhailov N.V., et al. Genomic analysis of virulence and antibiotic resistance of Klebsiella pneumoniae strains. Infection and Immunity. 2024;14(2):339–50. DOI: https://doi.org/10.15789/2220-7619-GAO-15645 EDN: https://elibrary.ru/cmtxuz
- Скачкова Т.С., Замятин М.Н., Орлова О.А. и др. Мониторинг метициллинрезистентных штаммов стафилококка в многопрофильном стационаре Москвы с помощью молекулярно-биологических методов. Эпидемиология и вакцинопрофилактика. 2021;20(1):44–50. Skachkova T.S., Zamyatin M.N., Orlova O.A., et al. Monitoring of methicillin-resistant staphylococcal strains in the Moscow medical and surgical center using molecular biological methods. Epidemiology and Vaccinal Prevention. 2021;20(1):44–50. DOI: https://doi.org/10.31631/2073-3046-2021-20-1-44-50 EDN: https://elibrary.ru/fwncis
- Якубцевич Р.Э., Лемеш А.В., Кирячков Ю.Ю. Патогенетические механизмы формирования генетической устойчивости к антибиотикам при лечении тяжелых инфекций в интенсивной терапии. Журнал Гродненского государственного медицинского университета. 2021;19(3):255–62. Yakubtsevich R.E., Lemesh A.V., Kiryachkov Yu.Yu. Pathogenetic mechanisms of formation of genetic resistance to antibiotics in the treatment of severe infections in intensive care. Journal of the Grodno State Medical University. 2021;19(3):255–62. DOI: https://doi.org/10.25298/2221-8785-2021-19-3-255-262 EDN: https://elibrary.ru/rtqhgv
- Malachowa N., DeLeo F.R. Mobile genetic elements of Staphylococcus aureus. Cell Mol. Life Sci. 2010;67(18):3057–71. DOI: https://doi.org/10.1007/s00018-010-0389-4
- Schwarz S., Cardoso M. Nucleotide sequence and phylogeny of a chloramphenicol acetyltransferase encoded by the plasmid pSCS7 from Staphylococcus aureus. Antimicrob. Agents Chemother. 1991;35(8):1551–6. DOI: https://doi.org/10.1128/aac.35.8.1551
Supplementary files

