


Production and purification of recombinant proteins VP2 and VP3 of the Alongshan virus of the Jingmenvirus group and evaluation of their immunochemical properties
- Authors: Bondarenko E.V.1, Ermolaeva E.A.1, Kholodilov I.S.1, Litov A.G.1
-
Affiliations:
- Chumakov Federal Scientific Center for Research and Development of Immune-and-Biological Products of Russian Academy of Sciences (Institute of Poliomyelitis)
- Issue: Vol 102, No 2 (2025)
- Pages: 213-222
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18829
- DOI: https://doi.org/10.36233/0372-9311-612
- EDN: https://elibrary.ru/ONPICA
- ID: 18829

Cite item

Abstract
Introduction. Alongshan virus is a representative of the unclassified group of Jingmenviruses (Flaviviridae), which is detected in Ixodes persulcatus, Ixodes ricinus ticks and various mosquito species in Russia, China, Finland and France. Unlike traditional orthoflaviviruses, the Alongshan virus genome is represented by 4 positive-sense RNA segments. The first and third segments of the genome encode proteins homologous to proteins of the replicative machinery of orthoflaviviruses, the remaining segments encode putative structural proteins that have no known homologues: segment 2 — VP1a (envelope protein), VP1b and NuORF; segment 4 — VP2 (capsid) and VP3 (membrane). Human cases of Alongshan virus-associated disease have been described.
The aim of this study is to develop a system for expression and purification of recombinant VP2 and hydrophilic site VP3 proteins to test their antigenic properties.
Materials and methods. Miass527 strain of Alongshan virus was used to produce hyperimmune mouse sera and recombinant proteins in a bacterial expression system. Bioinformatic analysis of sequences encoding target proteins and genetically engineered cloning were carried out in this study. Western blotting and enzyme immunoassay (ELISA) were performed to control the results.
Results. Recombinant proteins of Alongshan virus have been used in a laboratory diagnostic test system to determine the presence of antibodies to the virus. The obtained recombinant VP2 protein is able to detect antibodies in all tested sera of infected mice, as well as antibodies in human sera both in Western blotting and in enzyme immunoassay. At the same time, antibodies to the recombinant region of VP3 protein were detected irregularly in antiviral immune sera.
Conclusion. The detection of antibodies to Alongshan virus in patients confirms the necessity for further investigation of this group of viruses.
Full Text
Introduction
Members of the genus Orthoflavivirus (family Flaviviridae) infect vertebrates and invertebrates. Such important human pathogens as dengue virus, yellow fever virus, West Nile virus, tick-borne encephalitis virus (TBEV), Japanese encephalitis virus, etc. belong to this genus. Recently, a new group of viruses named Jingmenvirus, which is related to the Orthoflavivirus genus, has been characterized [1]. Unlike classical flaviviruses, Jingmenviruses have a segmented RNA genome [2]. The first and third segments of the Jingmenvirus genome encode proteins homologous to helicase and RNA-dependent RNA polymerase of orthoflaviviruses [3, 4]. The second and fourth segments encode unique proteins: envelope proteins VP1a and VP1b, presumably capsid protein VP2 and membrane protein VP3.
The Jingmenvirus group includes viruses such as Jingmen tick virus, Alongshan virus (ALSV), Yanggou tick virus and Takachi virus [1, 5, 6]. They have a wide geographical distribution and are detected in blood-sucking arthropods (especially ticks) and mammals, including those found in human sera [3, 7–13]. ALSV and Jingmen tick viruses apparently can cause acute infection in humans accompanied by fever [1, 14–16]. ALSV was first detected and isolated from the blood of a patient with febrile illness in China [17]. Later, ALSV was detected by polymerase chain reaction (PCR) in other patients: 86 people with a history of fever, headache, and tick bites between May and July 2017 were examined [17]. The virus was also detected in Ixodes persulcatus ticks and various mosquito species (Anopheles yatsushiroensis, Aedes vexans, Culex pipiens pallens and Culex tritaeniorhynchus) in China.
In Russia, the virus was detected in ticks in the Kaliningrad, Chelyabinsk and Ulyanovsk oblasts and in the Republics of Altai, Tatarstan, Karelia and Tyva [5, 18–20]. In connection with the described cases of human disease, epidemiological studies of Jingmenvirus group representatives are essential.
Currently, it is not known to which protein antibodies are produced during Jingmenvirus infection. Recombinant VP2 protein was successfully used to detect ALSV-specific antibodies in sheep and cattle in China by enzyme-linked immunosorbent assay (ELISA) [17]. ALSV-specific antibodies were detected in 9.2% (22/240) of the examined sheep and 4.6% (11/240) of the examined cattle. In 2019, Finnish scientists used constructs encoding the ALSV envelope proteins VP1a, VP1b, VP2 and VP3 transfected into Vero E6 mammalian cells. These cells were used to detect antibodies in the sera of 900 patients by immunofluorescence [21].
The aim of this study is to obtain recombinant proteins of ALSV and use them in a laboratory diagnostic test system to determine the presence of antibodies to the virus.
Materials and methods
Virus and cells
To obtain recombinant proteins, we used the Miass527 strain of ALSV isolated from I. persulcatus ticks collected in 2014 in Miass, Chelyabinsk region (NCBI access code: MN648770-MN648773) [1]. The recombinant sE TBEV protein was kindly provided by V.S. Baryshnikova [22].
Escherichia coli cells, strain TOP10 (Promega) were used for cloning, and strains JM109 and BL21 (Promega) were used for expression of recombinant proteins.
Production of hyperimmune blood sera
To obtain hyperimmune sera, mongrel mice ICR (Scientific Center of Biotechnology) were used, immunized under the skin with ALSV (strain Miass527) with Freund's adjuvant (BD) 3 times (once a week), 10 days after the last injection the blood was collected in total (decapitation). The obtained sera from 3 mice to ALSV were used in immunoblotting and ELISA. Serum from 1 mouse to TBEV (strain KE-328) was kindly provided by V.S. Baryshnikova [22]. Serum from an unimmunized mouse was used as a negative control.
The study protocol was approved by the Ethical Committee of the M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Products of the Russian Academy of Sciences (Poliomyelitis Institute) (Protocol No. 200923-1 of September 20, 2023).
Sera of the conditionally healthy population
Sera of conditionally healthy individuals with antibodies to TBEV from Moscow and Moscow region were kindly provided by the Center for Hygiene and Epidemiology in Moscow region.
Bioinformatics analysis
To determine signal peptides, hydrophilic and hydrophobic parts of proteins, the SignalP 4.1 Server1 program was used. DNA to Protein program on the Zbio.net website was used to determine protein mass.
Cloning of VP2 and VP3 protein genes of Alongshan virus (strain Miass527)
Reverse transcription was performed using Invitrogen SuperScript III reverse transcriptase (Thermo Fisher Scientifiс).
PCR was performed with a Veriti 96 Well Thermal Cycler (Applied Biosystems) using Platinum Super Fi II polymerase (Thermo Fisher Scientifiс).
PCR mix composition: SuperFi buffer 10 µl, 2.5 mM nucleotides 1 µl, primers (forward and reverse) 1 µl each, Platinum SuperFi II polymerase 1 µl, cDNA 2 µl, water 32 µl. Total volume 50 µl.
PCR program for Platinum Super Fi II polymerase is as follows:
- 98°С 30 s;
- 98°С 10 s;
- 60°С 10 s;
- 72°С 30 s/30 cycles;
- 72°С 5 min.
The following restriction endonucleases were used in this study:
BamHI and HindIII (Thermo Fisher Scientific) for plasmid pQE32 (Qiagen) and amplicons VP2 and VP3;
BamHI and XhoI (Thermo Fisher Scientifiс) for plasmid pet28a+ (plasmid collection of M.P. Chumakov Federal Scientific Center for Research and Development of Immunobiological Products of Russian Academy of Sciences) and the VP2 amplicon.
After restriction, the obtained vectors and inserts were run in 1% agarose gel (PanEco) based on 1xTBE buffer: 0.09 M Tris (DiaM), 0.09 M H3BO3 (Pushchino Laboratories), 2 mM EDTA (DiaM) and purified using centrifuge columns (Qiagen).
DNA concentration was measured by optical density on a NanoDrop One instrument (Thermo Fisher Scientifiс). The ligation reaction was performed using T4 DNA ligase (Thermo Fisher Scientifiс). The vector and insert were taken in a 5 : 1 ratio.
E. coli TOP10 cells were transformed with the obtained ligase mixtures by heat shock method and grown in LB medium (Table 1) at 37°C. The presence of the insert was verified by PCR with primers for cloning. The Sanger sequencing method was used to confirm the presence of polyhistidine-tag, start and stop codons, and the absence of nonsynonymous substitutions. Sequencing was performed using the BigDye Terminator v.3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) on an Applied Biosystems 3500 Genetic Analyzer (Waltham) instrument.
Transformation and cultivation of E. coli cells upon expression
During transformation, the following strains were used for protein expression: strain JM109 for plasmid pQE32 and strain Bl21 for pet28a+. LB, SOB and TB media were used for cell cultivation (Table 1).
Table 1. Composition of media for induction of expression of the target protein in E. coli JM109 and Bl21 cells
Medium | Ingredients | Concentration, g/L |
LB | Tripton | 10 |
Yeast extract | 5 | |
NaCl («Fluka») | 10 | |
SOB | Tripton | 20 |
Yeast extract | 5 | |
NaCl | 0,585 | |
KCl («Fluka») | 0,185 | |
TB | Tripton | 12 |
Yeast extract | 24 | |
Glycerol 99% | 20* |
Note. *Сoncentration of glycerol is given in mL/liter.
One cell colony was taken from a Petri dish and grown in 5 mL of medium supplemented with 100 ng/mL antibiotic (ampicillin for pQE32, kanamycin for pet28a+) for 18 hours. The cell suspension was then transferred to 250 mL of medium with 100 ng/mL ampicillin or 50 ng/mL kanamycin. When the cell mass reached an optical density of 0.5–0.8 at a wavelength of 600 nm (9.6 × 109 cells/mL), isopropyl-β-D-1-thiogalactopyranoside (IPTG; Helicon) was added. The cells were incubated at different temperatures and times with stirring. The cell mass was then precipitated by centrifugation at 1700g, 4°C for 30 min, and the resulting precipitate was washed with 50 mL of phosphate-salt buffer (PBS; Sigma-Aldrich).
The conditions under which recombinant proteins expression was performed, unless otherwise specified: incubation time of cells — 12 h in LB medium at 37°С and IPTG concentration 0.5 mM.
Cell disruption by ultrasound
The cell mass was resuspended in 20 mL of lysis buffer (HEPES 100 mM (DiaM), NaCl 0.15 M, pH 8.5) and treated with ultrasound (Soniprep 150 device, MSE) as follows: 3 times for 1 min each with a 7 ms pulse on ice. Then the cells were centrifuged (7800g, 4°C, 30 min), the precipitate was resuspended in 20 mL of lysis buffer with 8 M urea (to release the protein into the soluble fraction) and treated with ultrasound once for 30 s, followed by centrifugation (7800g, 4°C, 30 min).
Separation and purification of recombinant proteins
Recombinant proteins were purified by affinity chromatography using a ready-to-use Ni-NTA Fast Start kit (Qiagen).
Buffer change to PBS and concentration were performed using Amicon Ultra-15 10 kDa centrifuge ultrafilters (Merck).
The resulting recombinant proteins were separated by electrophoresis in 15% polyacrylamide gel (PAGE) under denaturing conditions (SDS-PAGE). A calibration plot constructed from known concentrations of BSA (Genesystool) was used to determine the concentration of target proteins. Further, the concentration of the target protein was measured using the GBox instrument (Syngene) in the GeneTools program (Syngene).
Obtaining negative control (Mock)
To obtain a negative control (Mock), the same manipulations were performed with pQE-32 and pet28a+ plasmids without the insert as with the constructs with the insert: transformation, cultivation of the corresponding bacterial cells, expression, isolation and purification. Controls were further used for visualization in PAGE, immunoblotting and ELISA.
Immunoblotting
The obtained recombinant proteins were separated by electrophoresis in 15% PAGE-SDS and transferred to nitrocellulose membrane (Bio-Rad). The membrane was incubated with 5% skimmed cow's milk (Best Value) in Tris-buffered saline (TBS: 25 mM Tris, 0.15 M NaCl, pH 8.3) for 1 h.
The membrane was then incubated with target serum for 1 h: mouse, human or to histidine tag. Next, the membrane was washed with TBS with 0.05% Tween-20 (TBS-T) and incubated with horseradish peroxidase (HRP)-labeled antibodies against mouse or human IgG, respectively (Abcam) for 1 h. If HRP-labeled antibodies against histidine tag (Abcam) were used, incubation with secondary labeled antibodies was not required.
Before imaging, the membrane was washed again with TBS-T, then imaging was performed using the ECL kit (Bio-Rad) in a Genesys gel-documentation system (Genesys).
Enzyme immunoassay
80 ng of protein diluted in PBS was added to a well of a 96-well plate and incubated overnight at 4°C. The plate was washed with PBS, incubated with 4% skimmed cow's milk (Best Value) in PBS for 1 h, then with mouse sera in PBS with 0.05% Tween-20 for 1 h. The plate was washed, incubated with HRP-conjugated antibodies against mouse IgG (Abcam) for 1 h, respectively, followed by washing and addition of TMB substrate (Sigma-Aldrich), after 30 min the reaction was stopped with 2 M sulfuric acid (Lenreactiv). The results were detected at a wavelength of 450 nm on a spectrophotometer (Thermo Fisher Scientific).
Selecting and obtaining targets for cloning
In the current study, the capsid protein VP2 was presumably chosen because Chinese scientists have already detected antibodies to this protein in cattle [13], as well as the membrane protein VP3, which was cloned for the first time in our study. The proteins are encoded in segment 4 of the ALSV genome.

Fig. 1. Schematic representation of VP2 and VP3 target regions chosen for cloning.
Black fragment – signal peptide of VP2 protein, blue – transmembrane domains of VP3 protein. 1 — VP3 protein region 1–267 aa, 2 — VP3 protein region 244–389 aa.
The sequence of the signal peptide (19 aa) was determined for the VP2 protein; it has no hydrophobic sites; the 243 aa fragment lacking the signal peptide was chosen for cloning because it could complicate the subsequent purification of the protein. VP3 protein contains 9 transmembrane hydrophobic domains, thus, hydrophilic regions 1–89 aa and 244–389 aa were chosen for cloning (Fig. 1). Presumed protein sizes were 25 kDa for VP2 is, and 10 and 18 kDa for hydrophilic regions of VP3-1 and VP3-2 proteins, respectively.

Fig. 2. Electrophoretic analysis of amplicons of target regions of VP2 and VP3 protein genes.
MW, DNA length marker. VP2, fragment encoding 1–89 aa; VP3, fragment encoding 244–389 aa VP3.
Then, based on the nucleotide sequences of VP2 protein and hydrophilic regions of VP3 protein, primers were selected (Table 2) and the corresponding PCR products were obtained (Fig. 2).
Table 2. Оligonucleotides for cloning the VP2 protein and the hydrophilic regions of the VP3 protein
Primer | Sequence | Genome regions based (GenBank #MN648773.2) |
VP2-28s | GAGCTAGGATCCAAGCCAAACGGAGCCCCAGAT | 168–188 |
VP2-28as | GAGCTACTCGAGCTACTGAAAAACCTGGTAGTTG | 857–872 |
VP3s 244–389 | GAGCTAGGATCCGACAAGGATCAAGCCTACCTC | 1576–1597 |
VP3as 244–389 | TAGCTCAAGCTTCCATTGGGTGTAGACCAGGT | 1998–2017 |
MiVP3s 1–89 | GCTAGGATCCGTGCGACCCCAACTACCAGGT | 848–868 |
MiVP3as 1–89 | TAGCTCAAGCTT TCTCTCCTCCAGTCGCC | 1095–1114 |
Obtaining vector constructions
The construct with the insertion of the 244-389 aa fragment of VP3 protein based on the pQE-32 vector was successful (Figs. 3, 4). However, with other inserts based on the pQE-32 vector, the expression of recombinant proteins either did not occur (in the case of the VP2 insert) or led to the death of bacterial cells at 3 h of cultivation (in the case of the insertion of the 1–267 aa fragment of VP3 protein), which indicates the possible toxicity of the protein for these bacterial cells. Therefore, we decided to assemble a new genetically engineered construct of pET28a+ plasmid with VP2 protein insertion (Fig. 3).

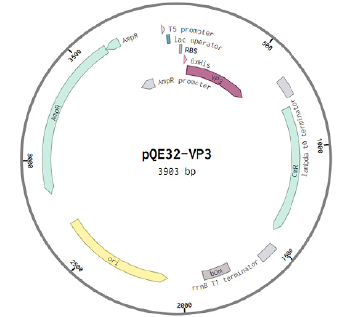
Fig. 3. Genetically engineered constructs which demonstrated expression of target proteins.
a — pQE-32 plasmid construct and VP3 protein fragment; b — pet28a plasmid construct and VP2 protein fragment. AmpR — ampicillin resistance gene; lac operator — lactose operator; bp — base pairs; KanR — kanamycin resistance gene; RBS — ribosome entry site; 6xHis — 6-histidine site; MCS — restriction sites.
The successful protein expression in pQE-32-244-389 and pet28a-VP2 constructs was confirmed using histidine-tag antibodies. Both recombinant proteins showed positive results in western blot (Fig. 4). Cells were cultured under the following conditions: LB medium, 37°C, IPTG concentration 0.5 mM, 12h.

Fig. 4. Immunoblotting results of recombinant VP3 (a) and VP2 (b) proteins with antibodies to histidine.
Optimization of conditions for recombinant protein expression
A number of experiments were undertaken to increase protein yield. The following parameters were optimized: cell culture medium, duration of incubation with IPTG, IPTG concentration and cell growth temperature. Fig. 5 shows that the highest concentration of VP3 target protein was achieved when 0.5 mM IPTG was added during bacterial growth for 12 h at 37°C. The use of different LB, SOB and TB media did not affect the expression of the partial recombinant VP3 protein (Fig. 5, c).

Fig. 5. PAGE-electrophoretic analysis of partial recombinant VP3 244-389 aa protein expressed at different IPTG concentrations (a), time (b), medium (c) and temperature (d).
In this regard, we determined the working conditions of target protein expression – cell cultivation for 12 h with the concentration of lactose operator inducer 0.5 mM at 37°C in LB medium.
Changes in the medium conditions, IPTG concentration and temperature did not affect the expression of recombinant VP2 protein in E. coli cells of strain BL21 (Fig. 6), but with increasing time of bacterial cell growth the expression increased.

Fig. 6. PAGE-electrophoretic analysis of partial recombinant VP2 protein expressed at different IPTG concentrations (a), time (b), medium (c) and temperature (d) of expression for the target protein.
Since the bacterial medium, growth temperature and IPTG concentration did not affect the expression of recombinant VP2 protein, we chose the working conditions for expression: culturing cells for 12 h at 37°C with a concentration of 0.5 mM lactose operator inducer in LB medium.
Purification of recombinant proteins
One of the important steps for further purification of proteins is to determine their solubility. For the partial recombinant VP3 protein, it was determined to be in the soluble fraction without urea (Fig. 7).

Fig. 7. Determination of VP2 (a) and VP3 (b) target protein fractions in PAGE.
a — cell lysate after sonication in lysis buffer: 1 — with 8 M urea; 2 — without additives; 3 — with 8 M urea and serine protease inhibitors; 4 — with serine protease inhibitors. b — cell lysate after 12 h of cultivation (1), Mock (2), cell lysate before IPTG addition (3), cell lysate after sonication in lysis buffer (4), precipitate fraction with 8 M urea (5)
The target VP2 protein is in the insoluble fraction (with 8 M urea), which could make it difficult to further obtain and purify. The addition of serine protease inhibitors, which may affect the solubility of the protein, was not successful — urea addition is necessary for the purification of recombinant VP2 protein.

Fig. 8. Purification of target protein VP2 in PAGE using Qiagen Ni-NTA Fast Start kit (a) and desalting with amicones (b).
a: 1 — VP2 protein in lysis buffer with 8 M urea; 2 — flow-through fraction; 3 — wash-buffer column washing fraction; 4 — eluate containing target protein; 5 — eluate containing protein degradation products.
b — recombinant VP2 protein and its degradation products (label: triangle); K+ — positive control.
The recombinant proteins were further purified on a gravity column by affinity chromatography (Fig. 8). After desalting and concentration on centrifuge filters, the concentration of proteins was measured by the Bradford method: for the partial recombinant VP3 protein it was 70 μg/mL, for VP2 protein — 120 μg/mL (when the proteins were expressed in 250 mL of medium with cell mass).

Fig. 9. Results of immunoblotting of target proteins with serum of mouse immunized with ALSV (strain Miass527).
a — expression of recombinant VP3 protein; b — cell lysate after expression of recombinant VP2 protein (1), isolated and desalted recombinant VP2 protein (2), Mock (3). HRP-labeled antibodies against mouse IgG (Abcam) were used for detection.
The ability of the recombinant VP2 protein to interact with antiviral antibodies was demonstrated by Western blot and the use of hyperimmune mouse sera to ALSV. Three hyperimmune mouse sera were tested — the VP2 protein interacted with all of them, while the partial recombinant VP3 protein was detected with only one. As an example, Fig. 9 shows an immunoblot of recombinant VP2 and VP3 proteins with hyperimmune mouse serum. Serum from an unimmunized mouse was used as negative serum in the Western blot.

Fig. 10. Results of immunoblotting of the target protein with hyperimmune mouse serum to TBEV (strain KE-328).
HRP-labeled antibodies against mouse IgG (Abcam) were used for detection. 1 — isolated and desalted recombinant VP2 protein; 2 — Mock; 3 — lysate of SPEV cells without TBE virus; 4 — recombinant sE protein with the size of 44 kDa [22].
To prove the specificity of recombinant VP2 protein in Western blot, mouse sera to TBEV were also used (Fig. 10). Recombinant TBEV sE protein served as a positive control [22]. The VP2 protein we obtained did not bind to TBEV antibodies.

Fig. 11. Analysis of mouse hyperimmune serum to ALSV Miass527 with recombinant VP2 protein (a) and Mock (b).
The wells of a 96-well plate were sensitized with VP2 protein solution; hyperimmune mouse serum to ALSV (C+) and serum from unimmunized mouse (C–) with the dilutions indicated in the figure were used in the assay. HRP-labeled antibodies against mouse IgG (Abcam) were used for detection.
The results of Western blot analysis were confirmed in ELISA with hyperimmune mouse serum to ALSV (Fig. 11). Different protein concentrations (20, 40, 80 and 120 ng/well) and serum dilutions (1 : 180 and 1 : 360) were tested in ELISA to determine the working concentration of recombinant proteins.
A protein concentration of 80 ng/well was found to be optimal. Serum from an unimmunized mouse was used as a negative control. To further confirm the results, the hyperimmune mouse serum to ALSV was also tested in ELISA, where a plasmid was used as a substrate, with which all the same manipulations were performed as with the insertion plasmid and recombinant protein during purification (Mock).
Positive ELISA and Western blot results allow the use of VP2 protein for detection of antibodies against ALSV in sera.
Analysis of sera from conventionally healthy subjects
Using the recombinant proteins obtained by us, 30 sera of conditionally healthy people from Moscow and Moscow region with antibodies to TBEV were tested. In ELISA analysis, samples of target protein at a concentration of 80 ng/well were used, Mock (which had the same concentration of total protein) was used as a negative control in the substrate, and serum of a conditionally healthy person who did not have antibodies to TBEV was used as a negative serum. Thus, 1 serum containing antibodies to the VP2 protein of ALSV was identified. No antibodies were detected to partial recombinant 244-389 aa VP3 protein.
To confirm the ELISA results, we performed a Western blot of the patient's ELISA-positive serum (Fig. 12).

Fig. 12. Results of immunoblotting of target protein with serum of conditionally healthy human. HRP-labeled antibodies against human IgG (Abcam) were used for detection.
Discussion
Previously, the VP2 protein was already obtained by Chinese scientists using the pET30a vector, which was expressed at 15°C in BL21 (DE3) E. coli cells [17]. In the current study, it was shown that VP2 protein was successfully expressed using plasmid pet28a(+), low temperature of cell cultivation was also tested, but it did not affect the expression level of recombinant protein. Efficient isolation of recombinant VP2 protein takes place upon addition of 8 M urea. At the same time, it was shown that the partial recombinant VP3 protein (vector pQE-32), which we obtained for the first time, is soluble. The yield of the target product is higher with the pet28a(+) vector than with pQE-32, and the obtained concentrations of both proteins are sufficient for multiple ELISAs.
Recombinant VP2 protein was used to detect antibodies in cattle in China [17]. In the currently study, we confirmed that VP2 protein has antigenic properties in immunoblot and ELISA. For the first time, we showed that the recombinant VP2 peptide of ALSV strain Miass527 obtained by us has no antigenic cross-reaction with TBEV in Western blot. It has been shown that when mice are immunized with live ALSV, antibodies to the recombinant VP2 protein are produced more regularly and less regularly to the partial recombinant VP3 protein. This may be due to the different spectrum of antibodies in the mouse sera obtained – the VP2 protein interacts with a large spectrum of antibodies produced at different stages of infection. The ELISA also detected antibodies to recombinant VP2 peptide in a conditionally healthy person with a history of tick bite, while no antibodies to recombinant VP3 peptide were detected in human sera.
1 SignalP 4.1 Server». URL: http://www.cbs.dtu.dk/services/TMHMM-2.0
About the authors
Ekaterina V. Bondarenko
Chumakov Federal Scientific Center for Research and Development of Immune-and-Biological Products of Russian Academy of Sciences (Institute of Poliomyelitis)
Author for correspondence.
Email: bondarenko_ev@chumakovs.su
ORCID iD: 0000-0002-5972-0761
junior researcher, Laboratory of biochemistry
Russian Federation, MoscowElena A. Ermolaeva
Chumakov Federal Scientific Center for Research and Development of Immune-and-Biological Products of Russian Academy of Sciences (Institute of Poliomyelitis)
Email: le.ermolaeva@mail.ru
ORCID iD: 0000-0002-0406-0604
junior researcher, Laboratory of biochemistry
Russian Federation, Moscow
Ivan S. Kholodilov
Chumakov Federal Scientific Center for Research and Development of Immune-and-Biological Products of Russian Academy of Sciences (Institute of Poliomyelitis)
Email: ivan-kholodilov@bk.ru
ORCID iD: 0000-0002-3764-7081
Cand. Sci. (Med.), leading researcher, Laboratory of arboviruses biology
Russian Federation, MoscowAleksander G. Litov
Chumakov Federal Scientific Center for Research and Development of Immune-and-Biological Products of Russian Academy of Sciences (Institute of Poliomyelitis)
Email: novosti-wxo@yandex.ru
ORCID iD: 0000-0002-6086-3655
Cand. Sci. (Biol.), leading researcher, Laboratory of arboviruses biology
Russian Federation, MoscowReferences
- Wang Z.D., Wang B., Wei F., et al. A new segmented virus associated with human febrile illness in China. N. Engl. J. Med. 2019;380(22):2116–25. DOI: https://doi.org/10.1056/NEJMoa1805068
- Ackermann M., Padmanabhan R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 2001;276(43):39926–37. DOI: https://doi.org/10.1074/jbc.M104248200
- Webster C.L., Waldron F.M., Robertson S., et al. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol. 2015;13(7):e1002210. DOI: https://doi.org/10.1371/journal.pbio.1002210
- Qin X.C., Shi M., Tian J.H., et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA. 2014;111(18):6744–9. DOI: https://doi.org/10.1073/pnas.1324194111
- Kholodilov I.S., Litov A.G., Klimentov A.S., et al. Isolation and characterisation of Alongshan virus in Russia. Viruses. 2020;12(4):362. DOI: https://doi.org/10.3390/v12040362
- Kholodilov I.S., Belova O.A., Ivannikova A.Y., et al. Distribution and characterisation of tick-borne flavi-, flavi-like, and phenuiviruses in the Chelyabinsk region of Russia. Viruses. 2022;14(12):2699. DOI: https://doi.org/10.3390/v14122699
- Maruyama S.R., Castro-Jorge L.A., Ribeiro J.M., et al. Characterisation of divergent flavivirus NS3 and NS5 protein sequences detected in Rhipicephalus microplus ticks from Brazil. Mem. Inst. Oswaldo Cruz. 2014;109(1):38–50. DOI: https://doi.org/10.1590/0074-0276130166
- Vandegrift K.J., Kapoor A. The ecology of new constituents of the tick virome and their relevance to public health. Viruses. 2019;11(6):529. DOI: https://doi.org/10.3390/v11060529
- Ladner J.T., Wiley M.R., Beitzel B., et al. A multicomponent animal virus isolated from mosquitoes. Cell Host Microbe. 2016;20(3):357–67. DOI: https://doi.org/10.1016/j.chom.2016.07.011
- Kobayashi D., Kuwata R., Kimura T., et al. Detection of jingmenviruses in Japan with evidence of vertical transmission in ticks. Viruses. 2021;13(12):2547. DOI: https://doi.org/10.3390/v13122547
- Temmam S., Bigot T., Chrétien D., et al. Insights into the host range, genetic diversity, and geographical distribution of jingmenviruses. mSphere. 2019;4(6):e00645–19. DOI: https://doi.org/10.1128/mSphere.00645-19
- Dinçer E., Hacıoğlu S., Kar S., et al. Survey and characterization of Jingmen tick virus variants. Viruses. 2019;11(11):1071. DOI: https://doi.org/10.3390/v11111071
- Pascoal J.O., Siqueira S.M., Maia R.D.C., et al. Detection and molecular characterization of Mogiana tick virus (MGTV) in Rhipicephalus microplus collected from cattle in a savannah area, Uberlândia, Brazil. Ticks Tick Borne Dis. 2019;10(1):162–5. DOI: https://doi.org/10.1016/j.ttbdis.2018.10.002
- Терновой В.А., Гладышева А.В., Семенцова А.О. и др. Обнаружение РНК нового многокомпонентного вируса у больных Крымской-Конго геморрагической лихорадкой на юге России. Вестник Российской академии медицинских наук. 2020;75(2):129–34. Ternovoi V.A., Gladysheva A.V., Sementsova A.O., et al. Detection of the RNA for new multicomponent virus in patients with Crimean-Congo hemorrhagic fever in southern Russia. Annals of the Russian Academy of Medical Sciences. 2020;75(2):129–34. DOI: https://doi.org/10.15690/vramn1192, EDN: https://elibrary.ru/jfodao
- Taniguchi S. Detection of Jingmen tick virus in human patient specimens: emergence of a new tick-borne human disease? EBioMedicine. 2019;43:18–9. DOI: https://doi.org/10.1016/j.ebiom.2019.04.034
- Jia N., Liu H.B., Ni X.B., et al. Emergence of human infection with Jingmen tick virus in China: a retrospective study. EBioMedicine. 2019;43:317–24. DOI: https://doi.org/10.1016/j.ebiom.2019.04.004
- Wang Z.D., Wang W., Wang N.N., et al. Prevalence of the emerging novel Alongshan virus infection in sheep and cattle in Inner Mongolia, northeastern China. Parasit. Vectors. 2019;12(1):450. DOI: https://doi.org/10.1186/s13071-019-3707-1
- Карташов М.Ю., Кривошеина Е.И., Курушина В.Ю. и др. Встречаемость и генетическое разнообразие вируса Алонгшан (Flaviviridae), выявленного в клещах на юге Восточной Сибири. Вопросы вирусологии. 2024;69(2):151–61. Kartashov M.Yu., Krivosheina E.I., Kurushina V.Yu., et al. Prevalence and genetic diversity of the Alongshan virus (Flaviviridae) circulating in ticks in the south of Eastern Siberia. Problems of Virology. 2024;69(2):151–61. DOI: https://doi.org/10.36233/0507-4088-223, EDN: https://elibrary.ru/jwvfoe
- Morozkin E.S., Makenov M.T., Zhurenkova O.B., et al. Integrated Jingmenvirus polymerase gene in Ixodes ricinus genome. Viruses. 2022;14(9):1908. DOI: https://doi.org/10.3390/v14091908
- Kholodilov I.S., Belova O.A., Morozkin E.S., et al. Geographical and tick-dependent distribution of flavi-like Alongshan and Yanggou tick viruses in Russia. Viruses. 2021;13(3):458. DOI: https://doi.org/10.3390/v13030458
- Kuivanen S., Levanov L., Kareinen L., et al. Detection of novel tick-borne pathogen, Alongshan virus, in Ixodes ricinus ticks, south-eastern Finland, 2019. Euro. Surveill. 2019;24(27):1900394. DOI: https://doi.org/10.2807/1560-7917.ES.2019.24.27.1900394
- Барышникова В.С., Турченко Ю.В., Шишова А.А. и др. Рекомбинантный гликопротеин Е вируса клещевого энцефалита для создания дифференцирующей тест-системы. Биотехнология. 2022;38(6):73–83. Baryshnikova V.S., Turchenko Yu.V., Shishova А.А., et al. Recombinant glycoprotein E of tick-borne encephalitis virus for a developing differentiated test system. Biotekhnologiya. 2022;38(6):73–83. DOI: https://doi.org/10.56304/S0234275822060023, EDN: https://elibrary.ru/hzkfvy
Supplementary files

