


Геномное разнообразие и анализ детерминант резистентности Salmonella enterica подвид enterica серовар Kentucky, изолированных в России
- Авторы: Кулешов К.В.1, Павлова А.С.1, Кремлева А.А.2, Карпенко А.Е.1, Михайлова Ю.В.1, Крутова Н.Е.1, Лисицына М.Р.1, Попова К.Р.1, Веселова О.А.1, Подколзин А.Т.1, Акимкин В.Г.1
-
Учреждения:
- Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
- Федеральный центр охраны здоровья животных
- Выпуск: Том 101, № 3 (2024)
- Страницы: 303-314
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://microbiol.crie.ru/jour/article/view/18523
- DOI: https://doi.org/10.36233/0372-9311-488
- EDN: https://elibrary.ru/owlgtw
- ID: 18523

Цитировать

Аннотация
Введение. Salmonella Kentucky сиквенс-типа ST198 относится к одному из эпидемиологически значимых клонов нетифоидных сальмонелл во всём мире и характеризуется наличием высокорезистентных штаммов, а также возможностью адаптации к различным животным-хозяевам и условиям окружающей среды.
Цель работы — исследование штаммов S. Kentucky, выделенных из различных источников на территории России, в аспекте их филогенетического положения в масштабах глобального разнообразия патогена и определение их генетических особенностей.
Материалы и методы. Методом полногеномного секвенирования исследовано 55 штаммов S. Kentucky, которые были изолированы в 2010–2022 гг. из различных источников (клинические штаммы, пищевые продукты, а также от животных, кормов и объектов окружающей среды). Полногеномное секвенирование проводили с использованием платформ «Illumina». Филогенетический анализ на основе анализа нуклеотидных вариаций, дополнительно включал 390 штаммов S. Kentucky.
Результаты. Бóльшая часть российских штаммов (n = 50) относилась к ST198, 4 штамма — к ST314, 1 штамм — к ST152. Из 50 российских штаммов ST198 44 принадлежали к международной монофилитической MDR-линии S. Kentucky ST198 и относились к 4 отдельным сублиниям, 6 штаммов занимали базальное положение по отношению к MDR-линии. Всего было выявлено 320 генов и мутаций, ответственных за резистентность к противомикробным препаратам. Наиболее часто встречались точечные мутации в области QRDR. В большинстве случаев для российских штаммов было характерно присутствие вариантов геномного острова SGI1-K. При этом гипотетическая структура SGI1 соотносилась с филогенетической кластеризацией сублиний S. Kentucky.
Выводы. Результаты исследования позволили оценить популяционную структуру российских штаммов S. Kentucky ST198 в мировом масштабе и определить генетические детерминанты антибиотикорезистентности, включая структуру геномного острова SGI1.
Ключевые слова
Полный текст
Введение
Одним из эпидемиологически значимых сероваров нетифоидных сальмонелл является Salmonella enterica подвид enterica серовар Kentucky (далее S. Kentucky). В ряде исследований описаны случаи изоляции штаммов, продуцирующих β-лактамазы расширенного спектра действия (БЛРС), а в некоторых случаях даже устойчивых к карбапенемам. S. Kentucky считается одним из целевых сероваров для мониторинга распространённости в птицеводстве [1].
Различные исследования последних лет проливают свет на возникновение, движущие силы и потенциальные угрозы серовара S. Kentucky, в частности, сиквенс-типа (ST) S. Kentucky ST198, который легко адаптируется к давлению отбора, оказываемому применением антибиотиков в различных условиях окружающей среды. Этот серовар сальмонелл эволюционировал от отсутствия устойчивости до 1990 г. к увеличению распространённости устойчивых к ципрофлоксацину изолятов в начале XXI в. (с 55% в 2007 г. до 88% в 2017 г. [2, 3]). В первые годы этого десятилетия в исследованиях регистрируют накопление генетических элементов, ответственных за устойчивость к широкому спектру противомикробных препаратов [2]. Интенсивное использование антибиотиков в животноводстве и медицине является одной из первостепенных причин этого явления. Глобализация торговли и возможности для путешествий создают благоприятные условия для распространения устойчивых бактериальных клонов на международном уровне. В Европе инфицирование людей штаммом S. Kentucky ST198 с множественной лекарственной устойчивостью (Multiple drug resistance, MDR) раньше главным образом ассоциировалось с поездками в Северную Африку или Юго-Восточную Азию, причём наиболее вероятным источником этих бактериальных изолятов указывался преимущественно Египет [2, 4, 5].
Показано, что все MDR-штаммы S. Kentucky ST198 принадлежат к одной генетической линии, накопившей детерминанты устойчивости к противомикробным препаратам с начала 1990-х гг. [5]. Различные детерминанты хромосомной устойчивости с середины 1990-х гг. связаны с интеграцией геномного острова 1 (Salmonella genomic island 1 — SGI1). SGI1 — геномный остров длиной 43 тыс. п.о., который первоначально описан у S. Typhimurium DT104 [6] и кодирует устойчивость к множеству противомикробных препаратов, включая амоксициллин, гентамицин и сульфонамиды [7]. За встраиванием SGI1 последовали кумулятивные мутации в генах gyrA и parC, что привело к устойчивости к налидиксовой кислоте, а затем к ципрофлоксацину в 2002 г. Предполагается, что именно за счёт особенностей генетической организации — хромосомо-интегрированной резистентности — и объясняется клональный успех этой MDR-линии и её быстрое и широкое распространение по всему миру [5, 8]. В предыдущих исследованиях продемонстрировано, что приобретение SGI1 в сравнении с приобретением плазмид резистентности не требует затрат на приспособленность (бактериальный фитнес) во время роста в условиях ограниченного количества питательных веществ [9], что может непосредственно влиять на эффективное распространение антибиотикорезистентных штаммов и заставляет внимательно отслеживать тенденции и динамику циркуляции этого патогена не только в региональном, но и в глобальном масштабе.
Существующие исследования на основе данных полногеномного секвенирования и анализа популяционной структуры послужили полезной основой для понимания продолжающейся эволюции MDR-линии S. Kentucky ST198 [5]. Штаммы MDR-линии до 2005 г. в основном обнаруживались в Египте и затем быстро распространились по Африке и Ближнему Востоку [4]. Ещё одним поводом для беспокойства является расширяющийся спектр источников обнаружения штаммов MDR-линии S. Kentucky ST198. Первоначально они обнаруживались у автохтонной домашней птицы, но затем у различных животных и в пищевых продуктах (контаминированные сальмонеллой специи во Франции и США), стадах индеек в Германии и Польше, диких животных) [4, 10–12].
Согласно данным по мониторингу за сальмонеллезами, S. Kentucky не входит в десяток доминирующих сероваров сальмонелл в России. Случаи обнаружения сальмонелл из различных источников не превышают 0,3% на протяжении последних лет1, что соответствует нескольким десяткам штаммов в год. При этом, согласно опубликованным данным, этот серовар не являлся этиологическим агентом случаев групповой заболеваемости с 2019 г. по настоящее время [13], а информация о выделении штаммов S. Kentucky ST198 в России ограничивается единичными случаями [14]. Вместе с тем вопрос о структуре популяции российских штаммов и их генетической характеристике сравнительно со штаммами, циркулирующими в других регионах мира, остаётся открытым.
Цель работы — исследование штаммов S. Kentucky, выделенных из различных источников в России, в аспекте их филогенетического положения в масштабах глобального разнообразия патогена и определения их генетических особенностей.
Материалы и методы
Отбор изолятов и микробиологические исследования
В период с 2010 по 2022 г. проведено исследование 55 штаммов S. Kentucky, выделенных на территории регионов Российской Федерации из различных источников.
Микробиологический анализ штаммов сальмонелл
Перед проведением процедуры экстракции тотальной ДНК для полногеномного секвенирования проводили рассев бактериальной культуры до единичных колоний и подтверждение серовара с использованием поликлональных («ПЕТСАЛ») и моноклональных («Sifin») сывороток.
Полногеномное секвенирование
Тотальную ДНК из 7 × 109 КОЕ экстрагировали с использованием набора «Рибо-преп» («Амплисенс»). Геномные библиотеки для полногеномного секвенирования каждого штамма сальмонелл готовили из 70 нг тотальной ДНК с использованием набора «NexteraXT» («Illumina»). Массовое параллельное секвенирование проводили на платформах MiSeq, Hiseq, NextSeq («Illumina»).
Обработка данных полногеномного секвенирования и de novo сборка геномов
Удаление адаптеров, нуклеотидных прочтений низкого качества проводили с использованием пакета BBTools2. De novo cборку контигов для каждого из штаммов проводили с помощью ассемблера SKESA [15]. Количество нуклеотидных прочтений и качество сборки считали достаточным для дальнейшего анализа при соблюдении следующих условий: (1) если была достигнута в среднем более чем 30-кратная глубина секвенирования при обратном картировании исходных нуклеотидных прочтений на собранные контиги; (2) соблюдались требования к метрикам полученных контигов3: общая длина контигов от 4 до 5,8 млн п.о, N50 более 20 тыс. п.о., число контигов — менее 600, доля нуклеотидов «N» — менее 3%, процент контигов, относящихся к роду Salmonella, — более 70%. На завершающем этапе валидации полученные контиги использовали для подтверждения принадлежности секвенированного штамма к серовару S. Kentucky c помощью программ SISTR [16] и SeqSero [17].
Филогенетический анализ
Всего коллекция геномов для филогенетического анализа включала 445 геномов штаммов S. Kentucky, из них 55 штаммов российского происхождения (номера доступа в GenBank SAMN42109132–SAMN42109186) и 390 штаммов, секвенированных в предыдущих исследованиях [5, 9, 18, 19]. Биоинформатический анализ проводили на основе поиска информативных однонуклеотидных полиморфизмов (SNP) путём картирования нуклеотидных прочтений каждого из секвенированных бактериальных изолятов на референс-геном S. Kentucky (номер доступа в NCBI: CP028357) с использованием программного конвейера SnapperDB [20]. Анализ проводили со следующими параметрами: минимальная глубина консенсуса — 10х, минимальное качество картирования прочтения для учёта вариации — 30, минимальная доля мажорного варианта — 90%, минимальное требуемое среднее покрытие по всему геному — 30х. Полученные профили SNP были конвертированы в формат выравнивания. Последующую реконструкцию филогенетического дерева и удаления SNP, расположенных в областях рекомбинаций, проводили в программе «Gubbins v. 3.2.1» [21]. Для подтверждения топологии дерева проводили бутстреп-анализ с 1000 повторениями. Филогенетические сублинии (генетически однородные группы профилей SNP) определяли с использованием программы «Fastbaps» с параметрами «optimise.baps». В результате использовали кластеры, идентифицированные на первом уровне кластеризации [22].
Определение сиквенс-типа, генетических детерминант антибиотикорезистентности и репликонов плазмид
ST на основе 7 генов домашнего хозяйства определяли с использованием программы mlst4. Поиск генов и точечных мутаций антибиотикорезистентности проводили с использованием «AMRFinderPlus v. 3.10.40» с параметрами «-I 0.9 –c 0.6 –O Salmonella» (минимальный процент идентичности — 90%, минимальное перекрытие — 60% с фильтрацией результатов, характерных для рода Salmonella) [23]. Определения типа плазмид проводили с помощью программы «MOB-suite v. 3.0.0» [24].
Анализ SGI1
Установление наличия и анализ гипотетической структуры острова SGI1 у исследуемых штаммов сальмонелл проводили путём картирования коротких нуклеотидных прочтений каждого из геномов на референс-последовательность SGI1-K (номер доступа в базе NCBI AY463797.8). В выборку для сравнения мы также включили геномы S. Kentucky, которые характеризовались различными вариантами острова SGI1-K, а также другими производными вариантами острова SGI1-K — SGI1-P и SGI-Q [5].
Результаты
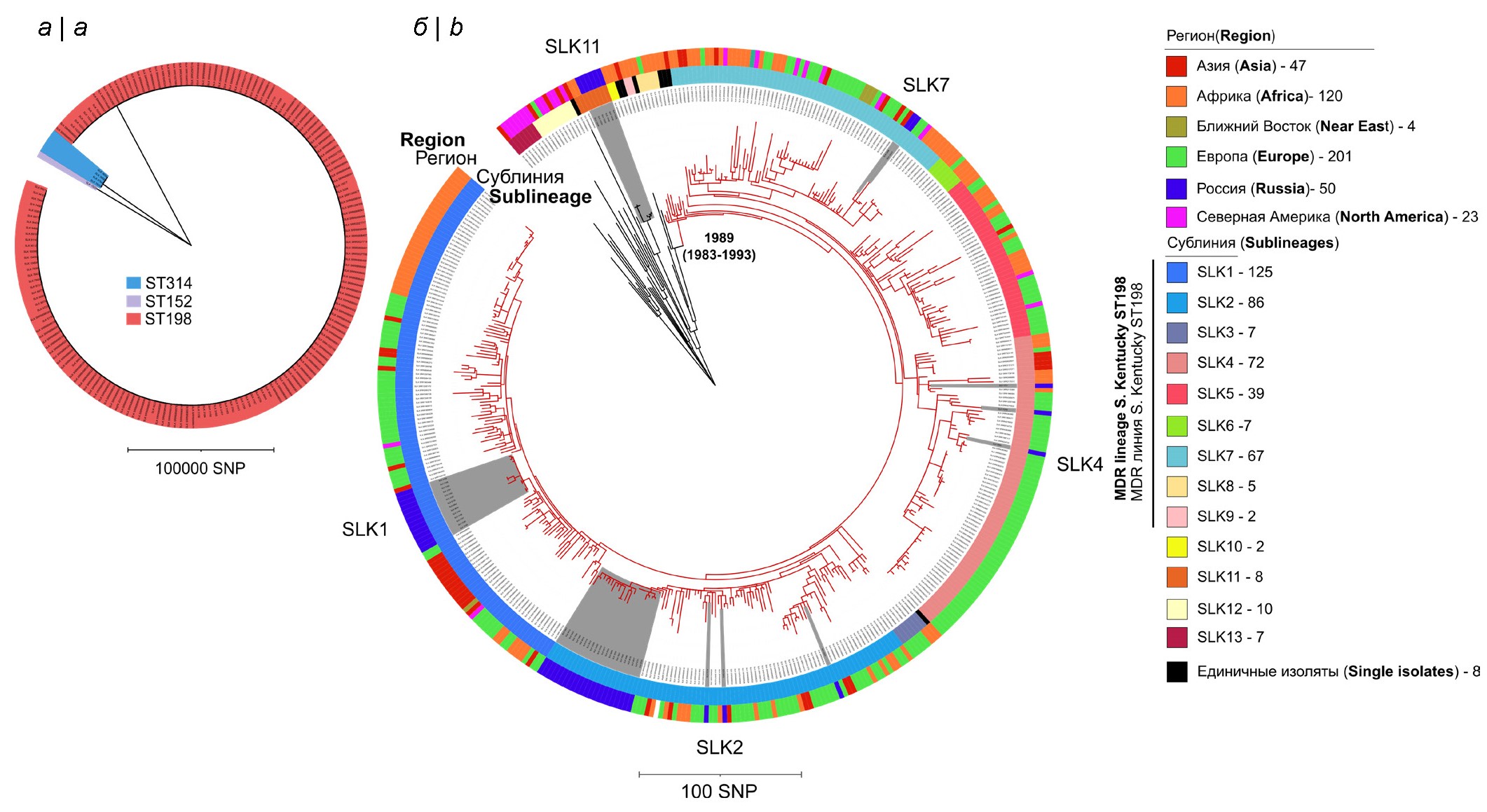
Согласно результатам анализа основная часть штаммов (n = 50) относилась к ST198 (90,9%), 4 (7,3%) штамма были представлены ST314 и только 1 (1,8%) штамм — ST152. Штаммы с этими ST принадлежали к отдельным филогенетическим линиям с высоким уровнем генетической дивергенции на уровне профилей коровых SNP, что свидетельствовало о полифилетической природе этого серовара, т.е. предковые штаммы этих ST эволюционно приобрели одни и те же О-антигены путём конвергентной эволюции (рис. 1, а) [25]. Поскольку основная часть российских штаммов принадлежала к генотипу ST198, дальнейший анализ был сосредоточен на детальном анализе этой группы. В качестве выборки для сравнения мы использовали ранее секвенированные геномы S. Kentucky ST198, которые депонированы в международных базах данных (n = 390). Эти штаммы характеризовались широким временным интервалом (1937–2022 гг.), географическим диапазоном получения штаммов (5 континентов), а также относились к разнообразным источникам выделения (человек, пищевые продукты, животные, окружающая среда).

Рис. 1. Филогенетическое дерево, реконструированное методом максимального правдоподобия.
а — дерево построено на основе SNP 173 штаммов S. Kentucky ST314, ST152 и ST198. Штаммы, относящиеся к отдельным сиквенс-типам, принадлежат к отдельным филогенетическим линиям с высоким уровнем генетической дивергенции;
б — дерево построено на основе SNP 445 штаммов S. Kentucky ST198. В качестве аутгруппы использовали геном штамма 98K (номер доступа SRR6898532), изолированный в США в 1937 г. от курицы. Красным выделена клада которая относится к MDR-линии S. Kentucky ST198, включает 9 сублиний SLK1-SLK9, определённых в программе «Fastbaps». Дата происхождения MDR-линии (1989 г.) указана в соответствии с публикацией J. Hawkey и соавт. [5]. Внешние сектора разного цвета представляют географические регионы, на территориях которых были изолированы штаммы. Серыми секторами на дереве выделены 50 штаммов S. Kentucky ST198, изолированных в России.
Fig. 1. Мaximum likelihood phylogenetic tree.
а — tree reconstructed based on SNPs of 173 S. Kentucky strains of sequence types ST314, ST152, and ST198. Strains belonging to individual sequence types belong to separate phylogenetic lineages with a high level of genetic divergence;
b — maximum likelihood phylogenetic tree reconstructed based on SNPs of 445 S. Kentucky sequence-type ST198 strains. The genome of strain 98K (accession number SRR6898532), isolated in the USA in 1937 from chicken, was used as an outgroup. The clade shown in red indicates the MDR lineage S. Kentucky ST198, which includes nine sublineages SLK1-SLK9, identified in the Fastbaps program. The date of MDR lineage origin (1989 year) is indicated according to J. Hawkey et al., 2019 [5]. The outer sectors of different colors represent the geographic regions in which the strains were isolated. Gray sectors on the tree highlight 50 strains of S. Kentucky ST198 isolated in Russia.
Филогенетический анализ 440 штаммов ST198 позволил определить 13 сублиний (генетических кластеров BAPS), представленных более чем 1 штаммом. При этом 9 сублиний относились к международной монофилитической MDR-линии S. Kentucky ST198, а 4 занимали базальное положение по отношению к ней (рис. 1, б). Базальные клады каждой сублинии международного MDR-клона включали штаммы, выделенные в странах Африки, что подтверждает гипотезу о возникновении MDR-линии S. Kentucky ST198 на африканском континенте и последующем распространении по всему миру.
Из 50 российских штаммов ST198 44 штамма принадлежали к международной монофилитической MDR-линии S. Kentucky ST198 и относились к 4 отдельным сублиниям (SLK-1, SLK-2, SLK-4, SLK-7), а 6 штаммов относились к сублинии SLK-11 и занимали базальное положение по отношению к MDR-линии S. Kentucky ST198 (рис. 1, б). Наибольшее число российских штаммов S. Kentucky (n = 25) относилось к сублинии SLK-2, наименьшее — к сублинии SLK-7 (n = 2). Отметим, что сублиния SLK-4 была в основном представлена штаммами из Европы.
Филогенетический анализ и географический регион выделения штаммов S. Kentucky на территории России не выявил корреляций. Вместе с тем для российских штаммов сублинии SLK-2 характерна преимущественная изоляция на территории североазиатской части России (Омская и Иркутская области, Республика Бурятия, Красноярский и Алтайский края), в то время как для сублинии SLK-11 — на территории европейской части России (Московская и Тульская области), а штаммы, относящиеся к сублиниям SLK-1, SLK-7 и SLK-4, обнаруживались на обеих территориях.
Анализ генетической кластеризации российских штаммов S. Kentucky на уровне различий в пределах 5 SNP позволил определить 6 t5-SNP-кластеров, которые относились к 3 сублиниям (рис. 2). Два t5-кластера были связаны с сублинией SLK-2. Один из t5-кластеров включал 8 штаммов, которые были выделены из различных источников (человек, окружающая среда и пищевой продукт) в течение 2019–2020 гг. Второй кластер включал 2 штамма, выделенных от человека в 2022 г.

Рис. 2. Генетическое разнообразие и популяционная структура российских штаммов S. Kentucky ST198 (n = 50).
Fig. 2. Genetic diversity and population structure of Russian strains of S. Kentucky ST198 (n = 50).
Филогенетическое дерево, реконструированное методом максимального правдоподобия на основе выравнивания SNP 51 штамма S. Kentucky ST198. В качестве аутгруппы использовали геном штамма S. Kentucky 93-6429 (номер доступа в архиве коротких прочтений SRR6898537), выделенный от человека в 1993 г. в Индонезии. Отдельные клады на филогенетическом дереве соответствуют выявленным генетическим группам российских изолятов S. Kentucky. В представленной схеме для каждого исследованного изолята отражены информация о регионах выделения штаммов; источниках; эпидемиологической ситуации, при которой изолированы штаммы (вспышка/спорадика); выявленные генетические кластеры на уровне различий в 5 SNP (t5-SNP-кластеры); информация о присутствии или отсутствии генетических детерминант антибиотикорезистентности и репликонов плазмид.
Maximum likelihood phylogenetic tree reconstructed based on SNP alignment of 51 S. Kentucky sequence-type ST198 strains. The genome of the S. Kentucky strain 93-6429 (short read archive accession number SRR6898537), isolated from a human in 1993 in Indonesia, was used as an outgroup. Individual clades on the phylogenetic tree correspond to the identified sublineages of Russian S. Kentucky isolates. The diagram shows isolate information: geographic region of isolation, source of strain, the epidemiological situation (outbreak/sporadic), identified genetic clusters at the level of differences in 5 SNPs (t5-SNP clusters), as well as information about the presence or the absence of genetic determinants of antibiotic resistance and plasmid replicons.
Российские штаммы S. Kentucky сублинии SLK-1 также включали 2 отдельных t5-SNP-кластера, к которым относилось 14 штаммов. Первый кластер включал 12 штаммов, выделенных из окружающей среды птицефермы и индейки во время энзоотии сальмонеллеза у птицы, тем самым подтверждая клональную связь случаев обнаружения сальмонелл на птицеферме. Второй кластер включал 2 спорадических штамма от человека в 2018 и 2019 гг.
Для сублинии SLK-11 были характерны 2 t5-SNP-кластера: 1 кластер — для штаммов, выделенных из комбикормов для индеек в 2020 г., другой — для штаммов, выделенных из помета индеек на птицеферме в 2022 г.
Генетические детерминанты антибиотикорезистентности и разнообразие плазмид S. Kentucky ST198
Все штаммы ST198, выделенные в России, содержали гены или мутации, ответственные за антибиотикорезистентность. При этом 62% штаммов содержали в общей сложности 6–11 генов резистентности. Всего среди 50 штаммов было выявлено 320 генов и мутаций, ответственных за резистентность к противомикробным препаратам. Наиболее часто встречались точечные мутации в области QRDR, определяющей устойчивость к хинолонам, а именно в генах gyrA (S83F) — 13,8% и parC (S80I) — 13,8%. Ген устойчивости к тетрациклинам tetA встречался у 11,9% штаммов. Гены blaTEM-1, aac(3)-Id и aadA7 присутствовали в 5% штаммов.
При анализе различных сочетаний генетических детерминант резистентности обнаружено 14 комбинаций. Наиболее часто встречаемой (26%) была комбинация gyrA(S83F, D87Y)–parC(S80I)–blaTEM-1–sul1-aac(3)-Id–aadA7–tet(A), 2-е и 3-е места по частоте встречаемости (по 8%) занимали комбинации gyrA(S83F, D87N)–parC(S80I)–sul1–tet(A) и gyrA(S83F, D87N)–parC(S80I)–dfrA14–sul1–sul2–aph(6)-Id–aph(3'')-Ib–tet(A).
Характер набора выявленных генов и мутаций резистентности в достаточной степени соответствовал филогенетической кластеризации изучаемых штаммов. Для всех штаммов сублинии SLK-2 было характерно наличие 3 хромосомных мутаций (gyrA (S83F, D87N) и parC (S80I)), а гены dfrA14, sul1, sul2, aph(6)-Id, aph(3’’)-Ib и tetA встречались у большей части штаммов. При этом в сравнении с другими в группе штаммов SLK-2 после 2015 г. отсутствовали гены bla и aadA.
Российские штаммы сублинии SLK-1 также несли 2 мутации в QRDR-регионе — gyrA(S83F) и parC(S80I), но отличались от сублинии SLK-2 мутацией в гене gyrA(D87Y). Все штаммы несли однотипный набор генов резистентности blaTEM-1–sul1–aac(3)-Id–aadA7–tet(A).
Для 2 штаммов сублинии SLK-7, как и для SLK-2, были характерны аналогичные мутации в генах gyrA (S83F, D87N) и parC (S80I). При этом штамм SLK_5298 включал весь набор генов, характерный для геномного острова SGI1-K blaTEM-1-sul1-aac(3)-Id-aph(6)-Id-aph(3'')-Ib-aadA7-tet(A) [19], а штамм SLK_10077 характеризовался отсутствием генов резистентности.
Штаммы сублинии SLK-4 характеризовались набором в gyrA (S83F, D87G) и parC (S80I). Более того, 2 штамма (SLK_10358 и SLK_4955) несли ген blaCTX-M-14, помимо других генов, кодирующих резистентность к антибиотикам.
В сравнении с остальными штаммами у представителей сублинии SLK-11 был выявлен только плазмид-опосредованный ген резистентности qnrB19. Мутаций в QRDR-регионе и других генов резистентности не выявлено.
Анализ разнообразия плазмид позволил определить наличие 13 известных типов несовместимости плазмид среди секвенированных российских изоля- тов. Наиболее часто встречались Col156, rep_cluster_ 2335 и rep_cluster_2350. Наибольшее разнообразие плазмид было характерно для штаммов сублинии SLK-2, которые, помимо вышеперечисленных плазмид, включали группы IncX, Inc-gamma, ColpVC.
Анализ острова SGI1
Присутствие в секвенированных геномах генов острова SGI1 рассматривалось как свидетельство встраивания SGI1 в хромосому бактерии, а именно в область с 3’-конца гена trmE до 5’-конца гена yidY. Результаты анализа перекрытия референсной последовательности SGI1-K для российских штаммов S. Kentucky представлены на рис. 3.

Рис. 3. Степень и характер перекрытия референсной последовательности SGI1-K (номер доступа в базе NCBI AY463797.8) короткими нуклеотидными прочтениями каждого из сравниваемых геномов.
Порядок штаммов в диаграмме соответствует порядку штаммов на филогенетическом дереве на рис. 2.
Fig. 3. The degree and nature of overlap of the reference sequence SGI1-K (NCBI accession number AY463797.8) with short nucleotide reads of each of the compared genomes.
The order of strains in the diagram corresponds to the order of strains in the phylogenetic tree in Fig. 2.
Основные отличия в вариантах SGI1-K заключаются в вариации не только состава генов острова, но и набора транспозонов. В частности, референтный SGI1-K несёт набор из 5 транспозонов Int4-Tn21-Tn1721-Tn5393-Tn3, в свою очередь ранее описанный вариант SGI1-K у штамма 08-KS6 характеризуется отсутствием транспозона Tn5393 и укороченными вариантом транспозона Tn1721 (Int4-Tn21-∆Tn1721-Tn3), у штамма 08-5707 укорочен транспозон Tn5393 (Int4-Tn21-Tn1721-∆Tn5393-Tn3), а у штамма 07-1511 присутствуют только 3 транспозона (Tn1721-Tn21-Tn3) с инверсией региона.
Штаммы сублинии SLK-11 характеризовались отсутствием как генов острова SGI1, так и мобильных генетических элементов Int4, Tn21, Tn1721, Tn3, свойственных SGI1-K, что свидетельствует об отсутствии интеграции острова SGI1 в хромосому.
Для штаммов сублинии SLK-2 перекрытие референс-последовательности SGI1-K варьировало от 19 до 61%. При этом большинство штаммов характеризовалось отсутствием основной части генов острова в диапазоне от гена S005 до гена resG и наличием транспозонов Int4, Tn21, Tn1721, несущих гены резистентности. В штаммах SLK_7836, SLK_7843 и SLK_7842, для которых отсутствовали мобильные элементы, было также характерно отсутствие генов антибиотикорезистентности. Штамм SLK_6643 характеризовался сходным характером перекрытия референс-последовательности с SGI1-Q. В свою очередь, штаммы SLK_5116 и SLK_4223 были аналогичны варианту SGI1-K 08-5707 с делецией коровых генов traG-resG и c набором мобильных элементов Int4-Tn21-Tn1721-Tn3.
Все штаммы сублинии SLK-1 имели аналогичный состав генов острова SGI1 и 90% перекрытия референс-последовательности SGI1-K. Набор мобильных элементов, включающий Int4-Tn21-Tn1721-Tn3, был аналогичен варианту SGI1-K, которые описан ранее в штамме 08-KS6.
В сублинии SLK-7 штамм SLK_10077 характеризовался 60% процентным перекрытием референтной последовательности за счёт наличия генов острова и отсутствием мобильных элементов, что коррелировало с отсутствием генов резистентности при анализе контигов. При этом нуклеотидные прочтения штамма SLK_5298 на 100% перекрывали референс-последовательность SGI1-K.
Штаммы сублинии SLK-4 характеризовались различающимися вариантами SGI1. Так, для штамма SLK_10358 (61% перекрытия) было характерно наличие всех генов острова, а из мобильных элементов присутствовал только транспозон Tn5393. Штаммы SLK_4955 (87% перекрытия) и SLK_1731 (85% перекрытия) характеризовались сходным набором транспозонов Int4-Tn21-Tn1721-Tn539. Однако в SLK_1731 отсутствовали гены острова SGI1 (S025-resG).
Обсуждение
Эпидемиологическая значимость серовара S. Kentucky обусловлена его потенциалом к распространению, приспособляемости к различным условиям окружающей среды и генетическими механизмами, включая хромосомный и плазмид- опосредованный, позволяющими реализовывать резистентность к широкому спектру противомикробных препаратов. Наряду с высокой долей штаммов, резистентных к ципрофлоксацину, зарегистрированы случаи комбинированной резистентности к ципрофлоксацину и цефотаксиму (цефалоспорины III поколения). В частности, штаммы с комбинированной устойчивостью были зарегистрированы на о. Мальта и относились к бактериальным изолятам, продуцирующим БЛРС [26]. Описаны также штаммы S. Kentucky с генами БЛРС (СTX-M-1, -M-14, -M-15, -M-104), цефалоспориназ AmpC β-лактазмаз (CMY-2 и -4) и карбапенемаз (VIM-2, OXA-48, NDM-1), локализованных не только на плазмидах, но и в хромосомном регионе SGI1 [4, 5, 19, 26–29]. Хотя сальмонеллез считается строго зоонозной инфекцией, высказываются предположения о распространении некоторых эпидемических клонов S. Kentucky от человека к человеку и о том, что человек является резервуаром [18].
К настоящему времени известны 10 различных ST S. Kentucky, 3 из которых (ST198, ST152 и ST314) встречаются наиболее часто [30]. В ряде случаев отмечается разная представленность этих ST — как территориально (межстрановые различия), так и относительно источников изоляции сальмонелл [31, 32]. Вместе с тем отмечено преобладание ST198 и ST152 у людей и у домашней птицы [33]. В нашем исследовании более 90% штаммов, ассоциированных с различными источниками выделения, относились к ST198, а ST152 был представлен единичным штаммом, что согласуется с международными данными.
Использование полногеномного секвенирования является золотым стандартом и универсальным инструментом геномного эпидемиологического надзора за социально значимыми патогенами [34, 35]. Возможность иерархической кластеризации филогенетически значимых маркеров определённого патогена на разных уровнях детализации и сравнения с последовательностями, депонированными в международных базах данных, не только даёт ценную информацию о тенденциях эволюции и оценке факторов распространения патогена в мировом масштабе, но и позволяет определить клонально-родственные связи между штаммами для эпидемиологического расследования вспышечной и спорадической заболеваемости [36–38].
Согласно проведённому филогенетическому анализу основная часть штаммов ST198, циркулирующих на территории России, относилась к 4 основным сублиниям международной монофилитической MDR-линии S. Kentucky ST198, которая включает штаммы с MDR-фенотипом к противомикробным препаратам [5]. Основная часть российских штаммов относилась к филогенетическим сублиниям SLK-1 и SLK-2. При этом длительный период изоляции штаммов из различных источников, относящихся к этим 2 сублиниям, может свидетельствовать о наличии 2 устоявшихся и генетически различающихся субпопуляций MDR-линии S. Kentucky ST198 на территории России. Интересно отметить, что характер филогенетической кластеризации на отдельные сублинии совпадал с определённой комбинацией из 3 мутаций в генах gyrA и parC, определяющей устойчивость к хинолонам, что позволяет использовать их в качестве маркеров для дифференциации российских штаммов S. Kentucky ST198 на основные филогенетические сублинии.
Два штамма сублинии SLK-4 — SLK_10358 (Иркутск, спорадический случай заболевания в 2022 г.) и SLK_4955 (репрезентативный штамм со вспышки сальмонеллеза в Ижевске в 2015 г.) — относились к представителям филогенетической сублинии, которая была ассоциирована с укоренившейся с 2005 г. в европейских странах популяцией S. Kentucky [18]. Особенностью этих ципрофлоксацин-резистентных штаммов S. Kentucky является наличие интегрированного в хромосому гена blaCTX-M-14, кодирующего БЛРС [18]. Этот ген также обнаруживался и в российских штаммах сублинии SLK-4. Случаи изоляции подобных штаммов могут указывать на единичные и независимые случаи заноса штаммов S. Kentucky blaCTX-M-14 с территории стран ЕС, однако это предположение требует исследования расширенной выборки штаммов.
Полученные данные, основанные на определении генетически близких штаммов, с использованием подхода кластеризации на уровне, не превышающем 5 нуклеотидных вариаций между штаммами (t5-SNP-кластер), выявил 6 групп клонально-родственных штаммов внутри 3 сублиний российских штаммов S. Kentucky ST198. При этом размер групп варьировал от 2 до 11 штаммов. В большинстве случаев выявленные t5-кластеры были связаны с циркуляцией определённого клона S. Kentucky ST198 на ограниченной территории и в относительно короткий промежуток времени (не более 1 года): при эпизоотической вспышке сальмонеллёза на птицеферме для индеек в Тульской области (2012 г.); выявлении штаммов сальмонелл в помёте индеек на птицеферме в Московской области (2022 г.); в комбикорме для индеек в (2020 г.); а также при 2 независимых случаях сальмонеллеза у людей в Ангарске и Иркутске в 2022 г. Вместе с тем наши исследования демонстрируют возможность существования скрытой циркуляции определённого клона S. Kentucky ST198 с выявлением штаммов в 2019 и 2020 гг. в Омске (n = 7) и Иркутске (n = 1), которые были ассоциированы со спорадическими случаями заболевания сальмонеллёзом и выделением сальмонелл из объектов окружающей среды и из пищевых продуктов, тем самым указывая на вероятную эпидемиологическую связь.
Применяемый в нашей работе подход к анализу структуры геномного острова SGI1 позволил нам опосредованно оценить его состав и продемонстрировать чувствительность данного геномного региона к генетическим перестройкам, вызываемым активностью транспозиционных элементов [5, 9, 19]. Эти перестройки могут привести к удалению некоторых или всех генов внутри SGI1 [5]. В большинстве случаев для российских штаммов характерно присутствие вариантов SGI1-K. Тем не менее в ряде геномов обнаруженный набор транспозонов отличался от существующих вариантов SGI1, что, вероятно, свидетельствует о присутствии нового варианта геномного острова. Высокая вариация этого острова также была отмечена в предыдущих исследованиях, где почти каждый штамм характеризовался различающимся по структуре SGI1. Помимо крупных делеций острова SGI1, некоторые штаммы имели инверсии всего или части сегмента, включающих гены устойчивости, а также перестановки транспозонов [5].
Анализ организации SGI1 и филогенетическая кластеризация штаммов на основе анализа профилей SNP в общих чертах соотносились друг с другом для различных сублиний SLK. Однако у штаммов SLK-2, принадлежащих к одному t5-SNP-кластеру, структура SGI1 демонстрировала отличия в наборе транспозонов. Подобные расхождения могут свидетельствовать о высокой скорости генетических перестроек региона SGI1 у клонально-родственных штаммов и объяснять разницу в наборе генов антибиотикорезистентности.
Присутствие SGI1 коррелировало с обнаруженным набором генов антибиотикорезистентности. Большинство штаммов (86%) характеризовались наличием генов aadA7, blaTEM-1, sul1 и tetA, которые, как известно, ассоциированы с SGI1 у штаммов S. Kentucky ST198 [19].
В совокупности выявленные гены устойчивости к противомикробным препаратам являлись ответственными за устойчивость к различным классам антибиотиков, включая аминогликозиды, β-лактамы, фениколы, хинолоны, сульфонамиды и тетрациклины. Основываясь на полученных данных, мы не можем уверенно говорить об ассоциации определённых генов резистентности с обнаруженным типом плазмид из-за отсутствия завершённой сборки генома. Однако полученные данные о разнообразии типов плазмид указывают на их способность к передаче генов устойчивости. Было показано, что малые плазмиды ColRNAI — «rep_cluster_2335» и «rep_cluster_2350» могут успешно нести разнообразный спектр генов резистентности, в частности, blaCTX-M-15, blaSHV-11, blaTEM-1B, blaOXA-1, ac(3)-IIa, strB, strA, aadA16, qnrB66, oqxA и oqxB [39–42].
Заключение
Несмотря на относительно небольшую выборку штаммов, нам удалось не только приблизиться к пониманию популяционной структуры российских штаммов S. Kentucky ST198 в мировом масштабе, но и провести детальное исследование генетических детерминант антибиотикорезистентности, включая структуру геномного острова SGI1. Полученные данные служат основой для понимания и отслеживания продолжающейся эволюции MDR-линии, которая представляет собой глобально распространённый клон, способный к быстрой экспансии и накоплению детерминант устойчивости к противомикробным препаратам. Наши данные демонстрируют случаи циркуляции клонально-родственных штаммов S. Kentucky ST198 в различных источниках, что свидетельствует о необходимости развития комплексного подхода в надзоре за сальмонеллёзами на основе концепции «Единое Здоровье».
1 Информационные бюллетени референс-центра по мониторингу за сальмонеллёзами № 35–26. URL: https://www.epid-oki.ru/otchety.html
2 BBTools. URL: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/ (дата обращения 12.12.2023)
3 EnteroBase. Quality Assessment evaluation. URL: https://enterobase.readthedocs.io/en/latest/pipelines/backend-pipeline-qaevaluation.html (дата обращения 12.12.2023).
4 mlst: scan contig files against traditional PubMLST typing schemes. URL: https://github.com/tseemann/mlst (дата обращения 12.12.2023).
Об авторах
Константин Валерьевич Кулешов
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Автор, ответственный за переписку.
Email: konstantinkul@gmail.com
ORCID iD: 0000-0002-5238-7900
к.б.н., зав. лаб. молекулярной диагностики и эпидемиологии кишечных инфекций
Россия, МоскваАнастасия Сергеевна Павлова
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: a.pavlova@cmd.su
ORCID iD: 0000-0003-4619-9337
н.с. лаб. молекулярной диагностики и эпидемиологии кишечных инфекций
Россия, МоскваАнна Александровна Кремлева
Федеральный центр охраны здоровья животных
Email: viktoriya1409@yandex.ru
ORCID iD: 0000-0002-6290-6639
н.с. отдела бактериологии Испытательной центральной научно-методической ветеринарной лаборатории
Россия, МоскваАнна Евгеньевна Карпенко
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: a.egorova@cmd.su
ORCID iD: 0000-0003-0486-1353
н.с. лаб. молекулярных механизмов антибиотикорезистентности
Россия, МоскваЮлия Владимировна Михайлова
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: mihailova@cmd.su
ORCID iD: 0000-0002-5646-538X
к.б.н., зав. лаб. молекулярных механизмов антибиотикорезистентности
Россия, МоскваНаталья Евгеньевна Крутова
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: nkrutova@cmd.su
ORCID iD: 0000-0003-2925-5376
м.н.с. лаб. молекулярной диагностики и эпидемиологии кишечных инфекций
Россия, МоскваМария Романовна Лисицына
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: kopylova@cmd.su
ORCID iD: 0009-0009-2344-6383
м.н.с. лаб. молекулярной диагностики и эпидемиологии кишечных инфекций
Россия, МоскваКристина Романовна Попова
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: c.bicmetowa@yandex.ru
ORCID iD: 0000-0003-3368-7833
м.н.с. лаб. молекулярной диагностики и эпидемиологии кишечных инфекций
Россия, МоскваОльга Александровна Веселова
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: sapienscri@gmail.com
ORCID iD: 0000-0002-5041-4370
н.с. лаб. молекулярной диагностики и эпидемиологии кишечных инфекций
Россия, МоскваАлександр Тихонович Подколзин
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: apodkolzin@pcr.ru
ORCID iD: 0000-0002-0044-3341
д.м.н., зам. директора по эпидемиологиии
Россия, МоскваВасилий Геннадьевич Акимкин
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
д.м.н., профессор, академик РАН, директор
Россия, МоскваСписок литературы
- Koutsoumanis K., Allende A., Alvarez-Ordóñez A., et al. Salmonella control in poultry flocks and its public health impact. EFSA J. 2019;17(2):e05596. DOI: https://doi.org/10.2903/j.efsa.2019.5596
- Le Hello S., Harrois D., Bouchrif B., et al. Highly drug-resistant Salmonella enterica serotype Kentucky ST198-X1: a microbiological study. Lancet Infect. Dis. 2013;13(8):672–9. DOI: https://doi.org/10.1016/S1473-3099(13)70124-5
- Le Hello S., Weill F.X., Guibert V., et al. Early strains of multidrug-resistant Salmonella enterica serovar Kentucky sequence type 198 from Southeast Asia harbor Salmonella genomic island 1-J variants with a novel insertion sequence. Antimicrob. Agents Chemother. 2012;56(10):5096–102. DOI: https://doi.org/10.1128/AAC.00732-12
- Le Hello S., Hendriksen R.S., Doublet B., et al. International spread of an epidemic population of Salmonella enterica serotype Kentucky ST198 resistant to ciprofloxacin. J. Infect. Dis. 2011;204(5):675–84. DOI: https://doi.org/10.1093/infdis/jir409
- Hawkey J., Le Hello S., Doublet B., et al. Global phylogenomics of multidrug-resistant Salmonella enterica serotype Kentucky ST198. Microb. Genom. 2019;5(7):e000269. DOI: https://doi.org/10.1099/mgen.0.000269
- Bie L., Fang M., Li Z., et al. Identification and characterization of new resistance-conferring SGI1s (Salmonella genomic island 1) in Proteus mirabilis. Front. Microbiol. 2018;9:3172. DOI: https://doi.org/10.3389/fmicb.2018.03172
- Doublet B., Golding G.R., Mulvey M.R., Cloeckaert A. Secondary chromosomal attachment site and tandem integration of the mobilizable Salmonella genomic island 1. PLoS One. 2008; 3(4):e2060. DOI: https://doi.org/10.1371/journal.pone.0002060
- Le Hello S. Bekhit A., Granier S.A., et al. The global establishment of a highly-fluoroquinolone resistant Salmonella enterica serotype Kentucky ST198 strain. Front. Microbiol. 2013;4:395. DOI: https://doi.org/10.3389/fmicb.2013.00395
- Cohen E., Davidovich M., Rokney A., et al. Emergence of new variants of antibiotic resistance genomic islands among multidrug-resistant Salmonella enterica in poultry. Environ. Microbiol. 2020;22(1):413–32. DOI: https://doi.org/10.1111/1462-2920.14858
- Beutlich J., Guerra B., Schroeter A., et al. Highly ciprofloxacin resistant Salmonella enterica serovar Kentucky isolates in turkey meat and a human patient. Berl. Munch. Tierarztl. Wochenschr. 2012;125(3-4):89–95. (in German)
- Münch S., Braun P., Wernery U., et al. Prevalence, serovars, phage types, and antibiotic susceptibilities of Salmonella strains isolated from animals in the United Arab Emirates from 1996 to 2009. Trop. Anim. Health Prod. 2012;44(7):1725–38. DOI: https://doi.org/10.1007/s11250-012-0130-4
- Wasyl D., Hoszowski A. First isolation of ESBL-producing Salmonella and emergence of multiresistant Salmonella Kentucky in turkey in Poland. Food Res. Int. 2012;45(2):958–61. DOI: https://doi.org/10.1016/j.foodres.2011.07.024
- Павлова А.С., Кулешов К.В., Крутова Н.Е. и др. Характеристика антибиотикорезистентности нетифоидных сальмонелл, циркулирующих на территории Российской Федерации в период с 2019 по 2022 год. Журнал микробиологии, эпидемиологии и иммунобиологии. 2023;100(5):287–301. Pavlova A.S., Kuleshov K.V., Krutova N.E., et al. Characteristics of antibiotic resistance of non-typhoidal Salmonella circulating in the Russian Federation in the period from 2019 to 2022. Journal of Microbiology, Epidemiology and Immunobiology. 2023;100(5):287–301. DOI: https://doi.org/ https://doi.org/10.36233/0372-9311-451 EDN: https://elibrary.ru/tmxvam
- Кафтырева Л.А., Егорова С.А., Макарова М.А. Детекция международных клонов высокого риска Salmonella и Escherichia coli – возбудителей заболеваний, передающихся с пищевыми продуктами, в Российской Федерации. Инфекция и иммунитет. 2020;10(3):565–9. Kaftyreva L.A., Egorova S.A., Makarova M.A. Detection of international high-risk clones of food-borne pathogens Salmonella and Escherichia coli in the Russian Federation. Russian Journal of Infection and Immunity. 2020;10(3):565–9. DOI: https://doi.org/ https://doi.org/10.15789/2220-7619-DOI-150 EDN: https://elibrary.ru/iybhmg
- Souvorov A., Agarwala R., Lipman D.J. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018;19(1): 153. DOI: https://doi.org/10.1186/s13059-018-1540-z
- Yoshida C.E., Kruczkiewicz P., Laing C.R., et al. The Salmonella In Silico Typing Resource (SISTR): an open web-accessible tool for rapidly typing and subtyping draft salmonella genome assemblies. PLoS One. 2016;11(1):e0147101. DOI: https://doi.org/10.1371/journal.pone.0147101
- Zhang S., den Bakker H.C., Li S., et al. SeqSero2: rapid and improved Salmonella serotype determination using whole-genome sequencing data. Appl. Environ. Microbiol. 2019;85(23):e01746-19. DOI: https://doi.org/10.1128/AEM.01746-19
- Coipan C.E., Westrell T., van Hoek A.H.A.M., et al. Genomic epidemiology of emerging ESBL-producing Salmonella Kentucky bla CTX-M-14b in Europe. Emerg. Microbes Infect. 2020;9(1):2124–35. DOI: https://doi.org/10.1080/22221751.2020.1821582
- Mashe T., Thilliez G., Chaibva B.V., et al. Highly drug resistant clone of Salmonella Kentucky ST198 in clinical infections and poultry in Zimbabwe. npj Antimicrob. Resist. 2023;1(1):6. DOI: https://doi.org/10.1038/s44259-023-00003-6
- Dallman T., Ashton P., Schafer U., et al. SnapperDB: a database solution for routine sequencing analysis of bacterial isolates. Bioinformatics. 2018;34(17):3028–9. DOI: https://doi.org/10.1093/bioinformatics/bty212
- Croucher N.J., Page A.J., Connor T.R., et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43(3):e15. DOI: https://doi.org/10.1093/nar/gku1196
- Tonkin-Hill G., Lees J.A., Bentley S.D., et al. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019;47(11):5539–49. DOI: https://doi.org/10.1093/nar/gkz361
- Feldgarden M., Brover V., Gonzalez-Escalona N., et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021;11(1):12728. DOI: https://doi.org/10.1038/s41598-021-91456-0
- Robertson J., Nash J.H.E. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018;4(8):e000206. DOI: https://doi.org/10.1099/mgen.0.000206
- Timme R.E., Pettengill J.B., Allard M.W., et al. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol. Evol. 2013;5(11):2109–23. DOI: https://doi.org/10.1093/gbe/evt159
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017. EFSA J. 2019;17(2):e05598. DOI: https://doi.org/10.2903/j.efsa.2019.5598
- Lei C.W., Zhang Y., Wang X.C., et al. Draft genome sequence of a multidrug-resistant Salmonella enterica serotype Kentucky ST198 with chromosomal integration of blaCTX-M-14b isolated from a poultry slaughterhouse in China. J. Glob. Antimicrob. Resist. 2020;20:145–6. DOI: https://doi.org/10.1016/j.jgar.2019.12.006
- Chen H., Song J., Zeng X., et al. National prevalence of Salmonella enterica serotype Kentucky ST198 with high-level resistance to ciprofloxacin and extended-spectrum cephalosporins in China, 2013 to 2017. mSystems. 2021;6(1):e00935-20. DOI: https://doi.org/10.1128/mSystems.00935-20
- El Hage R., Losasso C., Longo A., et al. Whole-genome characterisation of TEM-1 and CMY-2 β-lactamase-producing Salmonella Kentucky ST198 in Lebanese broiler chain. J. Glob. Antimicrob. Resist. 2020;23:408–16. DOI: https://doi.org/10.1016/j.jgar.2020.11.002
- Achtman M., Wain J., Weill F.X., et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012;8(6):e1002776. DOI: https://doi.org/10.1371/journal.ppat.1002776
- Slowey R., Kim S.W., Prendergast D., et al. Genomic diversity and resistome profiles of Salmonella enterica subsp. enterica serovar Kentucky isolated from food and animal sources in Ireland. Zoonoses Public Health. 2022;69(1):1–12. DOI: https://doi.org/10.1111/zph.12884
- Wang S., Liao X., Xiong Z., et al. Characterization of the emerging multidrug-resistant Salmonella enterica serotype Kentucky ST314 in China. Zoonoses Public Health. 2021;68(6):622–9. DOI: https://doi.org/10.1111/zph.12850
- Soltys R.C., Sakomoto C.K., Oltean H.N., et al. High-resolution comparative genomics of Salmonella Kentucky aids source tracing and detection of ST198 and ST152 lineage-specific mutations. Front. Sustain. Food Syst. 2021;5:695368. DOI: https://doi.org/10.3389/fsufs.2021.695368
- Deng X., den Bakker H.C., Hendriksen R.S. Genomic epidemiology: whole-genome-sequencing-powered surveillance and outbreak investigation of foodborne bacterial pathogens. Annu. Rev. Food Sci. Technol. 2016;7:353–74. DOI: https://doi.org/10.1146/annurev-food-041715-033259
- Franz E., Gras L.M., Dallman T. Significance of whole genome sequencing for surveillance, source attribution and microbial risk assessment of foodborne pathogens. Curr. Opin. Food Sci. 2016;8:74–9. DOI: https://doi.org/10.1016/j.cofs.2016.04.004
- Li S., He Y., Mann D.A., Deng X. Global spread of Salmonella Enteritidis via centralized sourcing and international trade of poultry breeding stocks. Nat. Commun. 2021;12(1):5109. DOI: https://doi.org/10.1038/s41467-021-25319-7
- Pijnacker R., Dallman T.J., Tijsma A.S.L., et al. An international outbreak of Salmonella enterica serotype Enteritidis linked to eggs from Poland: a microbiological and epidemiological study. Lancet Infect. Dis. 2019;19(7):778–86. DOI: https://doi.org/10.1016/S1473-3099(19)30047-7
- Chattaway M.A., Dallman T.J., Larkin L., et al. The transformation of reference microbiology methods and surveillance for Salmonella with the use of whole genome sequencing in England and Wales. Front. Public Health. 2019;7:317. DOI: https://doi.org/10.3389/fpubh.2019.00317
- Agyepong N., Govinden U., Owusu-Ofori A., et al. Genomic characterization of multidrug-resistant ESBL-producing Klebsiella pneumoniae isolated from a Ghanaian teaching hospital. Int. J. Infect. Dis. 2019;85:117–23. DOI: https://doi.org/10.1016/j.ijid.2019.05.025
- Lerminiaux N., Mitchell R., Bartoszko J., et al. Plasmid genomic epidemiology of blaKPC carbapenemase-producing Enterobacterales in Canada, 2010–2021. Antimicrob. Agents Chemother. 2023;67(12):e0086023. DOI: https://doi.org/10.1128/aac.00860-23
- Nakamura K., Seto K., Lee K., et al. Global population structure, genomic diversity and carbohydrate fermentation characteristics of clonal complex 119 (CC119), an understudied Shiga toxin-producing E. coli (STEC) lineage including O165:H25 and O172:H25. Microb. Genom. 2023;9(3):mgen000959. DOI: https://doi.org/10.1099/mgen.0.000959
- Rehman M.A., Rempel H., Carrillo C.D., et al. Virulence genotype and phenotype of multiple antimicrobial-resistant Escherichia coli isolates from broilers assessed from a «One-Health» perspective. J. Food Prot. 2022;85(2):336–54. DOI: https://doi.org/10.4315/JFP-21-273
Дополнительные файлы

