


Production and characterization of chimeric Bst-like polymerases and their application in isothermal amplification combined with rapid RNA extraction methods using the example of the mumps virus
- Authors: Zamotaeva T.L.1, Dedyaeva E.A.1, Mikheeva O.O.1, Pika M.I.1, Cherkashin E.A.1, Cherkashina A.S.1, Akimkin V.G.1
-
Affiliations:
- Central Research Institute of Epidemiology
- Issue: Vol 102, No 4 (2025)
- Pages: 391-403
- Section: SCIENCE AND PRACTICE
- URL: https://microbiol.crie.ru/jour/article/view/18909
- DOI: https://doi.org/10.36233/0372-9311-604
- EDN: https://elibrary.ru/TRVYOB
- ID: 18909

Cite item

Abstract
Introduction. Bst polymerase plays a key role in the rapid diagnosis of infectious diseases due to its unique biochemical properties and potential application in loop-mediated isothermal amplification (LAMP). Several analogs of Bst polymerase have been described in the literature; however, these enzymes have not been widely used in molecular diagnostics.
The aim of the study is to obtain recombinant Bst and Btlv polymerases with the Sso7d domain and to test new possibilities for their application.
Materials and methods. Expression constructs carrying the polymerase gene were obtained using standard genetic engineering methods. The target enzyme was produced in Escherichia coli cells. Purification was carried out using metal-affinity chromatography methods followed by dialysis and concentration. RNA-dependent DNA polymerase (reverse transcriptase) and DNA polymerase activities of the enzymes were determined using non-radioactive methods with fluorescent detection. The functional properties of the enzymes were assessed using the Amplisens SARS-CoV-2-IT reagent kit and a method designed for the detection of mumps virus RNA in biological material using the LAMP format combined with reverse transcription.
Results. In the E. coli-based expression system, the following recombinant chimeric enzymes with displacing activity have been obtained: Bst_Sso7d, Bst_Sso7d_mut4 and Btlv_Sso7d. The developed cultivation and purification protocols allow for the production of enzymes in soluble form with a yield of up to 25% of the collected cell mass. Functional testing showed that in LAMP, the chimeric polymerases demonstrated similar activity to Bst polymerase without the Sso7d domain. At the same time, the Btlv_Sso7d polymerase exhibited increased reverse transcriptase activity and resistance to inhibitors.
Conclusion. The obtained chimeric polymerase Btlv_Sso7d, due to its improved properties, can be used in reagent kits for the diagnosis of infectious diseases by the LAMP method when using nucleic acid extraction methods.
Full Text
Introduction
Reducing the time of research with the help of rapid tests is one of the key trends in laboratory diagnostics. This approach is particularly relevant for increasing the throughput of the laboratory in the context of mass screenings and allows medical personnel or epidemiologists to perform diagnostics in resource-limited settings, which contributes to providing timely assistance to patients, quickly identifying infected individuals, promptly investigating infectious outbreaks, and taking appropriate epidemiological measures, as well as preventing the excessive prescription of preventive and therapeutic measures. Such rapid tests must meet certain requirements such as high accuracy and speed, simplicity and accessibility, as well as high stability during storage and transportation [1, 2].
The coronavirus pandemic has spurred the development of isothermal amplification methods. The loop-mediated isothermal amplification (LAMP) method [3–5] has high sensitivity and specificity, and an important advantage is that the amplification reaction occurs at a constant temperature (without thermal cycling). This allows for research to be conducted both in equipped clinical diagnostic laboratories and in field conditions where specialized laboratory equipment is not available. The key role in the further development of reagent kits for the express diagnosis of infectious diseases using the LAMP method belongs to Bst polymerase [6, 7] and its modifications. Bst polymerase is a large fragment of DNA polymerase I (Bst-LF), isolated from the Geobacillus stearothermophilus thermophilic bacterium (formerly known as Bacillus stearothermophilus) [8] and having an optimal temperature of 60–70°C. The enzyme was isolated by J. Stenesh et al. in 1972 [9], four years before the discovery of Taq polymerase [10, 11]. Subsequently, both for research purposes and in production solutions, recombinant enzymes, particularly Bst polymerase, cloned and expressed in Escherichia coli bacterial cells, have generally been used. The bacterial system based on E. coli cells is characterized by simplicity and low cultivation costs, high microbial growth rates, and a wide range of different vectors for recombinant protein expression have been developed for it. Approaches to cloning and obtaining recombinant enzymes in a bacterial expression system based on E. coli cells have been described for Bst polymerase and similar polymerases with displacing activity from other organisms [12–14]. Since the specific catalytic activity of the enzyme is influenced, among other factors, by the characteristics of the nucleotide sequence of the gene, expression conditions, as well as the extraction and purification protocol, all these stages require optimization when obtaining any enzyme.
The main approaches to modifying the physicochemical characteristics of an enzyme according to the practical objectives of the user are directed mutagenesis and the addition of protein domains with specified properties [15, 16]. Such modifications allow for an increased yield of soluble enzyme during expression in E. coli cells, simplify the purification process, and also produce enzymes with improved properties such as higher activity and thermostability, as well as resistance to salts and inhibitors. The unmodified Bst polymerase has insufficient processivity because, in the native organism, it is primarily involved in DNA repair [17]. The modified enzyme exhibits much greater processivity, which is due to the presence of auxiliary proteins in the cells that enhance the stability of the polymerase-DNA complex. The Sso7d protein belongs to the family of DNA-binding proteins isolated from the Sulfolobus genus archaeon, and is stable over a wide range of temperatures and pH. Several authors describe a strategy of fusing polymerases with the Sso7d protein or similar proteins (Sto7d, SSB, TBD, DBD) to obtain chimeric enzymes with increased processivity, displacing activity, thermostability and tolerance to inhibitors, including urea, whole blood and NaCl [18–22].
Thermophilic bacteria of related species can develop different survival strategies, which are due, among other things, to differences in the properties of their enzymes. In this regard, potentially interesting directions include the cloning and obtaining of recombinant DNA polymerases from new sources, such as a related organism and the closest homolog — the Geobacillus thermoleovorans thermophilic bacterium (formerly known as Bacillus thermoleovorans) [23, 24].
The aim of this study was to obtain chimeric Bst-like polymerases from G. stearothermophilus and to compare them with the homolog Btlv polymerase from G. thermoleovorans to evaluate their potential application in isothermal amplification reactions combined with rapid RNA extraction methods.
Materials and methods
Obtaining the Bst_Sso7d, Bst_Sso7d_mut4, Btlv_Sso7d genes
The nucleotide sequence encoding the amino acid sequence of Btlv polymerase from G. thermoleovorans was obtained by the assembly method using long overlapping primers—the staircase method [25]. Restriction sites were introduced at the ends of the nucleotide sequence: NdeI at the 5' end and XhoI at the 3' end for subsequent re-cloning into the pET16b+ expression vector. As a result, the pET16-Btlv-Nhis expression vector was obtained. The correctness of the nucleotide sequence of the cloned gene was confirmed by sequencing.
As sources of the Bst and Bst_mut4 genes, plasmids previously obtained in the laboratory were used: pET16_Bst and pET16_Bst_NHis_m4, respectively [7, 26]. The template for amplifying the Sso7d gene was also the previously obtained construct pPSS, which contains the wild-type Sso7d gene.
To obtain the genes of chimeric enzymes, amplification of the target enzyme gene and the Sso7d gene of the DNA-binding domain was carried out. The obtained amplicons were extracted and purified from the gel and ligated together using flanking primers (Table 1).
Table 1. Primer sequences used for gene cloning
Matrix | Name | 5’-3’ sequence | PCR2 |
Bst_Sso7d | |||
рЕТ16_Bst | BstF | gaaaggaggaggagctctaacatctgcggaaggcgaaaaaccg | |
BstR | agtctcgagttatttcgcatcataccagg | ˅ | |
pPSS | SsoF | tcgtcatatggcgaccgtgaagttcaagtataaag | ˅ |
SsoR | agatgttagagctcctcctcctttcttctgtttttccag | ||
Bst_Sso7d_mut4 | |||
рЕТ16_Bst_NHis_m4 | BstF | gaaaggaggaggagctctaacatctgcggaaggcgaaaaaccg | |
BstR | agtctcgagttatttcgcatcataccagg | ˅ | |
pPSS | SsoF | tcgtcatatggcgaccgtgaagttcaagtataaag | ˅ |
SsoR | agatgttagagctcctcctcctttcttctgtttttccag | ||
Btlv_Sso7d | |||
pET16-Btlv-Nhis | BtlvF | gaaaggaggaggagctctaacatctccgtcttctgaggaagaaaagcc | |
BtlvR | aagtctcgagttatttcgcatcataccaagtagaaccgtagtg | ˅ | |
pPSS | SsoF | tcgtcatatggcgaccgtgaagttcaagtataaag | ˅ |
SsoR | agatgttagagctcctcctcctttcttctgtttttccag | ||
To obtain protruding A-ends, the purified amplicon was incubated for 30 minutes at 72°C in the presence of Taq polymerase and a mixture of deoxynucleotides (dNTPs). The target product was then cloned into the pGEM-T vector (Promega). The presence of the target sequence and its accuracy were confirmed by Sanger sequencing.
Obtaining expression plasmid vectors containing the Bst-Sso7d, Bst-mut4-Sso7d, Btlv-Sso7d genes
Plasmid DNA containing the gene of the chimeric enzyme was treated with the NdeI and XhoI restriction endonucleases, and the resulting restriction product was cloned into the pET16b+ plasmid vector, which had been pre-treated with the same restriction endonucleases. As a result, expression vectors containing genes that encode the following hybrid proteins were obtained: Bst_Sso7d with a molecular weight of 75 kDa, Bst_Sso7d_mut4 with a molecular weight of 75.2 kDa, and Btlv_Sso7d with a molecular weight of 75 kDa. The accuracy of the nucleotide sequence of the cloned genes was confirmed by sequencing.
Selection of E. coli strains for the expression of the Bst_Sso7d, Bst_Sso7d_mut4, and Btlv_Sso7d genes
As host strains for the constructed expression vectors pET16-Bst_Sso7d, pET16-Bst_Sso7d_mut4, and pET16-Btlv_Sso7d, the E. coli strains ER2566, BL21de3 pLys, and Rosetta De3 were used. Transformed cells were plated on LB medium (1% Bacto-tryptone, 0.5% yeast extract, 1% NaCl) with agar containing 100 µg/mL ampicillin for ER2566 cells and 20 µg/mL chloramphenicol for BL21 (DE3) pLys and Rosetta (DE3) cells, and incubated for 16 hours at 37°C to obtain individual colonies. Then, 7–8 colonies were transferred to 100 mL of LB medium with 100 µg/mL ampicillin and incubated for 18 hours at 37°C on a shaker at 180 rpm to obtain an overnight culture. The obtained overnight cultures of E. coli producer strains were transferred to LB medium with 100 µg/mL of ampicillin in Erlenmeyer flasks (the inoculation percentage was 2%) and incubated at 37°C with shaking at 160 rpm. When the optical density of the bacterial culture reached 0.8 optical units, isopropyl-β-D1-thiogalactopyranoside was added to a concentration of 0.4 mM, and the culture was incubated at 23°C and 37°C for 4 and 24 hours. The optical density was measured spectrophotometrically at a wavelength of 595 nm. Cell biomass was obtained by centrifugation for 20 minutes at 4°C and 4000 rpm on an Avanti JXN-30 (Beckman Coulter) centrifuge.
Isolation of Bst_Sso7d, Bst_Sso7d_mut4, and Btlv_Sso7d
The cell biomass (2 g) of E. coli BL21 (DE3)pLys/pET16-Bst_Sso7d, BL21 (DE3)pLys/pET16-Bst_Sso7d_mut4, and BL21 (DE3)pLys/pET16-Btlv_Sso7d producer strains was re-suspended in a buffer solution of 50 mM Tris-HCl, 100 mM NaCl, pH 8.5 with 1 mM PMSF at a ratio of 1:10 (w/v) and disrupted using a Branson sonifier 250 ultrasonic disintegrator (Branson Ultrasonics) for 20 minutes at 4°C (cycle — 0.5 s, amplitude — 50%). Then centrifuged at 8000 rpm for 30 minutes in an Allegra X-30R centrifuge (Beckman Coulter). After centrifugation, the supernatant was diluted 2-fold with a buffer solution of 50 mM Tris-HCl, 100 mM NaCl pH 8.5 and applied to the IMAC FF chromatographic sorbent, which had been pre-equilibrated with a buffer solution of 50 mM Tris-HCl, 100 mM NaCl pH 8.5 (buffer solution A). The removal of contaminating proteins was carried out with buffer solution A. The target protein was eluted with a linear gradient of buffer solution A with 500 mM imidazole.
After metal-chelate chromatography, the fractions containing the target protein were dialyzed against a buffer solution of 20 mM Tris, 100 mM NaCl, 0.5% Tween-20, 0.1 mM EDTA, pH 8.3. After dialysis, glycerol was added to the protein solution to a final concentration of 50%.
Determination of enzyme activity
The RNA-dependent DNA polymerase (reverse transcriptase) activity of the enzymes was determined using a non-radioactive method with fluorescent detection, based on the formation of a duplex of polyadenylated RNA and the oligonucleotide primer dT18, into which the GelStar intercalating dye (Lonza) was incorporated. The DNA polymerase activity of the enzymes was determined using a non-radioactive method [27]. An oligonucleotide containing a hairpin structure at the 3' end was used as the template. In the presence of Mg2+ ions, polymerases catalyzed the incorporation of deoxynucleotides, thereby extending the matrix. The described non-radioactive methods allow for the determination of enzyme activities using amplifiers with an optical module for real-time fluorescence detection.
The stability of the enzymes to temperature exposure was assessed using differential scanning fluorimetry in the range of 55–85°C at 1°C increments, with a step duration of 50 seconds. The melting curves were detected in the Fam channel on the CFX 96 device (Bio-Rad Laboratories).
The optimal ion concentrations in the reaction mixture were selected similarly to the analysis of polymerase activity using KCl concentrations in the range of 50–500 mM.
Analytical methods
The protein concentration was determined using the QuDye Protein kit (LLC LumiProbe RUS) on the Qubit 4 fluorimeter (Thermo Scientific), and the protein purity was assessed using SDS-PAGE electrophoresis under denaturing conditions [28]. When performing SDS-PAGE electrophoresis under denaturing conditions, a protein length marker — a molecular weight marker (ThermoScientific) — was used.
Loop-mediated isothermal amplification combined with reverse transcription
To assess the potential use of the obtained chimeric enzymes in new reagent kits for the diagnosis of infectious diseases, testing was conducted on the AmpliSens SARS-CoV-2-IT reagent kit (RU No. FSS 2021/14599). As samples, a genetic construct representing the MS2 bacteriophage with a specific insertion of the coronavirus ORF1ab gene was used. The reaction mixtures contained 0.8 µL of the tested enzymes at a concentration of 0.288 U/µL or the same amount of Bst polymerase without the Sso7d domain, as well as Reverse Transcriptase (MMLv). Each sample was amplified in 3 replicates on the Rotor-Gene Q device (Qiagen).
The functional properties of the enzymes were also tested in a laboratory method designed for detecting the RNA of the mumps virus in biological material using the LAMP format with combined reverse transcription. We amplified a fragment of the phosphoprotein gene of the mumps virus (amplicon size 225 base pairs, GC content 54%). Urine and oropharyngeal mucosal swabs were used as samples, and the presence of mumps virus RNA was confirmed by Sanger sequencing using the BigDye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific) on the 3500xL Genetic Analyzer (Thermo Fisher Scientific).
Nucleic acid extraction was performed using the EDEM express kit (RU No. 2010/07828), as well as a lysis solution containing guanidine hydrochloride. As a control extraction method, the RIBO-prep reagent kit (RU No. FSS 2008/03147) was used. The reaction mixtures contained oligonucleotides complementary to the target amplification sites, as well as an intercalating dye, which allowed for the registration of the accumulation of the specific amplification product by measuring the intensity of the fluorescent signal in real-time mode; as well as a mixture of glycerin with thioglycerol, salts, and surfactants (all additional reagents used were developed and produced at the Central Research Institute of Epidemiology of Rospotrebnadzor). As a control batch, the non-mutated form of Bst polymerase without the Sso7d domain was used at a protein concentration equal to that of the tested enzymes. The reaction was conducted according to the program: 37°C for 5 minutes, 65°C for 30 seconds, 40 cycles with detection in the FAM channel (total duration 25 minutes).
Assessment of enzyme stability to LAMP inhibitors
To assess the stability of LAMP enzymes against inhibitors, mucin (Sigma-Aldrich) was used, as well as pre-characterized residual samples of human biological material (urine, pharyngeal mucosa swabs, plasma, and whole blood). Swabs from the oropharyngeal mucosa were stored in a transport medium for the storage and transport of respiratory swabs (RU No. FSS 2009/05011; Central Research Institute of Epidemiology of Rospotrebnadzor).
Inhibitors were added to the reaction mixture for LAMP. The reaction was conducted according to the program: 37°C for 5 minutes, 65°C for 30 seconds, 40 cycles with detection in the FAM channel (total duration 25 minutes). The results were evaluated based on the presence/absence of the fluorescent signal (detected/not detected) and the values of the threshold cycles (Ct), which is sufficient for diagnostic systems with qualitative determination.
Results
The Sso7d family includes small, numerous, non-specific DNA-binding proteins, first discovered in the Archaea sulfolobus hyperthermophilic bacterium. They have a mass of 7–10 kDa and exhibit various types of functional activity: stabilization of the double helix, annealing of DNA at temperatures above its melting point, and prevention of protein aggregation. Moreover, such proteins alter the conformation of DNA, causing the unwinding of the DNA double helix [29].
To obtain recombinant Bst_mut4- and Btlv-polymerases, unique synthetic sequences were developed with consideration of codon usage optimization for expression in E. coli. Next, the Sso7d gene was attached to the Bst- and Btlv-polymerase genes, and these constructs were cloned into the pET16b+expression vector for prokaryotic expression in E. coli cells. During the selection of host strains for the expression of the genes Bst_Sso7d, Bst_Sso7d_mut4 and Btlv_Sso7d, the dynamics of enzyme accumulation were studied at different temperatures (23 and 37°C) and induction times for protein biosynthesis (4 and 24 hours). As a result, it was shown that all enzymes were effectively accumulated in a soluble form at 23°C for 24 hours in E. coli BL21 (DE3)pLys cells (Tables 2–4).
Table 2. Selection of host strains for the expression of Bst_Sso7d genes
E-coli host strain | Cultivation conditions | Protein content, % | Cell biomass extraction, g/L |
ER2566 | 23°С, 4 h | 10 | 2,1 |
23°С, 24 h | 15 | 2,3 | |
37°С, 4 h | 12 | 2,3 | |
37°С, 24 h | 12 | 2,5 | |
Rosetta De3 | 23°С, 4 h | 23 | 2,4 |
23°С, 24 h | 23 | 2,6 | |
37°С, 4 h | 21 | 2,5 | |
37°С, 24 h | 24 | 2,9 | |
BL21 De3 pLys | 23°С, 4 h | 23 | 2,7 |
23°С, 24 h | 25 | 3,14 | |
37°С, 4 h | 24 | 2,9 | |
37°С, 24 h | 21 | 3,2 |
Table 3. Selection of host strains for the expression of Bst_Sso7d_mut4 genes
E-coli host strain | Cultivation conditions | Protein content, % | Cell biomass extraction, g/L |
ER2566 | 23°С, 4 h | 10 | 2,1 |
23°С, 24 h | 11 | 2,5 | |
37°С, 4 h | 14 | 2,5 | |
37°С, 24 h | 16 | 1,7 | |
Rosetta De3 | 23°С, 4 h | 19 | 2,2 |
23°С, 24 h | 21 | 2,4 | |
37°С, 4 h | 21 | 2,3 | |
37°С, 24 h | 22 | 2,5 | |
BL21 De3 pLys | 23°С, 4 h | 21 | 2,1 |
23°С, 24 h | 23 | 2,6 | |
37°С, 4 h | 21 | 2,5 | |
37°С, 24 h | 20 | 2,7 |
Table 4. Selection of host strains for the expression of Btlv_Sso7d genes
E-coli host strain | Cultivation conditions | Protein content, % | Cell biomass extraction, g/L |
ER2566 | 23°С, 4 h | 18 | 2,0 |
23°С, 24 h | 16 | 2,5 | |
37°С, 4 h | 16 | 2,4 | |
37°С, 24 h | 14 | 2,8 | |
Rosetta De3 | 23°С, 4 h | 18 | 2,3 |
23°С, 24 h | 17 | 2,4 | |
37°С, 4 h | 19 | 2,5 | |
37°С, 24 h | 19 | 2,7 | |
BL21 De3 pLys | 23°С, 4 h | 21 | 2,4 |
23°С, 24 h | 23 | 2,5 | |
37°С, 4 h | 23 | 2,5 | |
37°С, 24 h | 21 | 2,7 |
For the purification of enzymes from cellular proteins, metal-affinity chromatography with a linear imidazole gradient was used (Fig. 1).

Fig. 1. Electrophoregram of purified enzymes Bst_Sso7d (a), Bst_Sso7d_mut4 (b), and Btlv_Sso7d (c).
M — molecular weight marker; 1 — clarified cell lysate; 2 — wash with metal-chelating resin; subsequent — fractions after affinity chromatography.
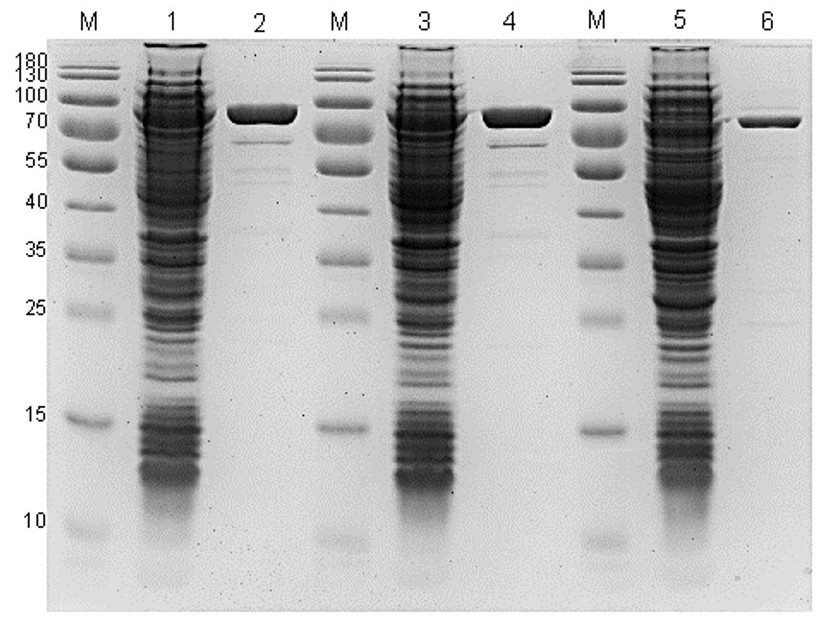
Fractions with a purity of over 90% were pooled and dialyzed against a buffer solution of 20 mM Tris, 100 mM NaCl, 0.5% Tween-20, 0.1 mM EDTA pH 8.3, followed by concentration. According to the electrophoretic analysis, the enzyme purity was at least 90% (Fig. 2) with a concentration of at least 2 mg/mL.

Fig. 2. Electropherogram of the purification of enzymes Bst_Sso7d_mut4, Bst_Sso7d, and Btlv_Sso7d.
M — molecular weight marker; 1, 3, 5 — clarified cell lysate of Bst_Sso7d_mut4, Bst_ Sso7d, and Btlv_Sso7d respectively; 2, 4, 6 — final purified enzyme preparations of Bst_Sso7d_mut4, Bst_ Sso7d, and Btlv_Sso7d respectively.
It is worth noting that, despite the higher percentage of protein content from the total mass of wet cells: in the case of Bst_Sso7d expression — 25% compared to 23% for Bst_Sso7d_mut4 and Btlv_Sso7d, after purification, the latter two enzymes showed higher yields. This is due to the fact that optimizing the codon composition of the coding sequence leads to an increase in the yield of the protein in a soluble form.
Testing the polymerase activity of the isolated enzymes Bst_Sso7d, Bst_Sso7d_mut4, and Btlv_Sso7d was conducted on samples of MS2 bacteriophage RNA with a specific insert containing a fragment of the SARS-CoV-2 genome, using the reverse transcription LAMP method with reagents from the AmpliSens SARS-CoV-2-IT kit. The concentration of the template was 105 copies per reaction. All new enzymes exhibited polymerase activity; however, the average threshold cycle values differed from the control batch of Bst polymerase without the Sso7d domain, with an increase in values by an average of 3.73 for Bst_Sso7d, 3.42 for Bst_Sso7d_mut4 and 2.88 for Btlv_Sso7d (Fig. 3), which may indicate suboptimal reaction conditions for the new enzymes.

Fig. 3. Evaluation of the activity of the obtained polymerases in the AmpliSens SARS-CoV-2-IT reagent kit (mean threshold cycle values).
All the studied enzymes showed comparable results in experiments assessing their resistance to temperature exposure (Fig. 4).

Fig. 4. Thermostability of chimeric polymerases.
a — thermal denaturation profiles; b — derivatives of fluorescence values with respect to temperature.
The isolated enzymes Bst_Sso7d, Bst_Sso7d_mut4, and the comparison enzyme exhibited weak RNA-dependent DNA polymerase (reverse transcriptase) activity both in the method [7] and when tested in the AmpliSens SARS-CoV-2-IT kit without the reverse transcriptase enzyme. At the same time, Btlv_Sso7d polymerase exhibited reverse transcriptase activity comparable to that of reverse transcriptase (MMLv), but only in the presence of KCl and (NH4)2SO4, which allows the enzyme to be used not only for DNA/cDNA amplification but also for RNA reverse transcription. Changing the composition of the reaction mixture (mainly the addition of KCl) allowed for an increase in the polymerase activity of all chimeric enzymes, with the optimal concentration of KCl in the buffer for LAMP being 200 mM (Fig. 5).

Fig. 5. Optimal KCl concentration for chimeric polymerases.
LAMP-based reagent kits allow for the reduction of amplification time from 1.5–2.5 hours (PCR method) to 25–40 minutes, while maintaining high specificity due to the use of 4–6 oligonucleotides. However, the process of nucleic acid extraction from biological material is still quite lengthy. The samples under investigation are treated with a lysis solution, resulting in the destruction of bacterial cell walls and viral envelopes, followed by the release of cellular components and nucleic acids into the solution. The subsequent wash steps in the extraction protocol allow for the removal of substances and cell components from the solution that inhibit PCR. Express methods are characterized by the absence of a washing step or the presence of only one washing step: clinical material taken in a special transport medium (for example, TS-EDEM) is subjected to thermal treatment and centrifugation, resulting in the precipitation of insoluble components, while the nucleic acids remaining in the supernatant are used for PCR. However, after disinfection without extraction or after express extraction, RNA/DNA samples contain impurities that can act as PCR inhibitors, such as components of transport media, lysis buffer, blood, swabs, urine and other biomaterials.
The functional properties of the polymerases obtained in the study were tested for resistance to the most commonly encountered inhibitors (components of whole blood, plasma, urine, mucin) in the LAMP reaction in a model system using positive control samples. Inhibitors were added to the reaction mixture containing primers from the AmpliSens SARS-CoV-2-IT reagent kit and isolated MS2 bacteriophage RNA with a specific insert containing a fragment of the SARS-CoV-2 genome. The results were evaluated based on the presence/absence of the fluorescent signal (detected/not detected), which is sufficient for diagnostic systems with qualitative determination (Table 5).
Table 5. Comparison of the effects of inhibitors on the detection of mumps virus RNA using the tested enzymes
Inhibitors | Detection of mumps virus RNA | ||||
Name | Concentration | Bst-polymerase (control) | Bst_Sso7d | Bst_Sso7d_mut4 | Btlv_Sso7d |
Whole blood, vol. % | 0 | + | + | + | + |
0,5 | + | + | + | + | |
1,0 | + | + | + | + | |
2,5 | + | – | – | + | |
5,0 | + | – | – | + | |
10,0 | – | – | – | + | |
Blood plasma, vol. % | 0 | + | + | + | + |
1 | – | – | – | + | |
Mucin, mg/ml | 0 | + | + | + | + |
0,2 | + | + | + | + | |
0,5 | + | + | + | + | |
Urine, vol. % | 0 | + | + | + | + |
5 | + | – | – | + | |
10 | – | – | – | + | |
Modified enzymes based on Bst polymerase, containing the DNA-binding domain Sso7D: Bst_Sso7d and Bst_Sso7d_mut4, demonstrated reduced stability in the presence of whole blood and urine in the reaction mixture compared to the control enzyme. At the same time, the Btlv_Sso7d enzyme is characterized by higher resistance to the presence of whole blood (10 vol. %), plasma (1 vol. %), and urine (10 vol. %). All enzymes retain their activity in the presence of mucin up to 0.5 mg/ml.
Further experiments to determine the effect of inhibitors on the efficiency of the LAMP reaction were conducted only using the Btlv_Sso7d enzyme. The resistance of the Btlv_Sso7d polymerase to inhibitors from biological material (mucus from oropharyngeal swabs, salts from urine), components of the transport medium (transport medium for storing and transporting respiratory swabs (RU No. FSS 2009/05011)), and the lysis solution (1M guanidine hydrochloride) was also tested on 12 clinical urine samples and 12 oropharyngeal mucosa swab samples containing mumps virus RNA at a concentration of 105–108 copies/mL, isolated by three different methods: a precipitation-based method using the RIBO-prep kit; the EDEM express method; treatment with a 1M guanidine hydrochloride solution (for disinfecting the biological material) without subsequent washes. All isolated samples were then compared using the LAMP method without the addition of reverse transcriptase (MMLV) (Fig. 6).

Fig. 6. Detection of the mumps virus in biological samples using LAMP with various polymerases without the addition of reverse transcriptase.
When using Btlv_Sso7d polymerase, unlike the control enzyme, the LAMP reaction maintains its efficiency under all studied conditions, and all biological samples containing mumps virus RNA used in the study are identified as positive. There is also a less significant increase in Ct values when analyzing samples after nucleic acid extraction using the EDEM express method, compared to the control enzyme. This confirms that the Btlv_Sso7d polymerase possesses sufficient reverse transcriptase activity and increased inhibitor tolerance for the qualitative detection of mumps virus RNA (Fig. 6).
Due to its higher tolerance to inhibitors, the use of the new Btlv_Sso7d polymerase in isothermal amplification reactions based on the LAMP method, combined with rapid nucleic acid extraction methods that include only the lysis stage, will expedite molecular diagnostics and make it possible to use LAMP-based tests in field conditions or at the bedside of a patient.
Discussion
The LAMP method is a promising direction in the molecular diagnosis of infectious diseases. The main advantage of the method is the shorter analysis time: the amplification stage takes only 25–40 minutes, while the sensitivity and specificity of the method are comparable to that of PCR [1, 2]. The isothermal reaction mode allows for the use of simpler equipment for conducting the reaction: a thermostat with a fluorescence detection module instead of an amplifier.
Besides the actual amplification stage, any analysis most often includes the nucleic acid extraction stage. The extraction of nucleic acids is carried out to eliminate the main amplification inhibitors that may be present in biological samples. On average, depending on the method used, the extraction stage can take from 1 to 3 hours. Reducing this stage by using express nucleic acid extraction methods (10–30 minutes) will significantly decrease the overall research time. For the successful implementation of such a strategy, the development and application of enzymes resistant to the main amplification inhibitors are necessary.
The Bst polymerases obtained in this study, containing the DNA-binding domain Sso7d: Bst_Sso7d and Bst_Sso7d_mut4, demonstrated reduced stability in the presence of whole blood and urine in the reaction mixture compared to the control enzyme without additional domains. At the same time, the Btlv_Sso7d enzyme is characterized by higher resistance to the presence of whole blood (10 vol. %), plasma (1 vol. %), and urine (10 vol. %). All enzymes retain their activity in the presence of mucin up to 0.5 mg/ml. These data demonstrate that the addition of the DNA-binding domain did not lead to increased resistance to the inhibitory effect of urine and whole blood in the case of Bst polymerase. Similar studies have been published in the literature, indicating the opposite effect when using various Bst-like polymerases [17, 18, 30, 31]. However, the related chimeric enzyme Btlv_Sso7d obtained in this work demonstrates high resistance to the inhibitory effect of whole blood, plasma and urine components on the LAMP reaction. Such a difference in results may be explained by the characteristics of the genetic engineering constructs, the presence or absence of additional spacers between the DNA-binding domain and the polymerase, the structure of these spacers, and the conditions of cultivation and purification of recombinant proteins. The obtained data demonstrate the need for further detailed studies in this area to determine the influence of DNA-binding domains on the properties of chimeric enzymes in each specific case and to establish the relationships between the structure and functions of chimeric enzymes.
Conclusion
In a bacterial expression system based on E. coli cells, recombinant chimeric enzymes with displacing activity have been obtained: Bst_Sso7d, Bst_Sso7d_mut4 and Btlv_Sso7d. The developed protocols for obtaining and purifying the enzymes allow for the production of soluble enzymes with a yield of up to 25% of the total collected cell mass. In LAMP reactions, chimeric polymerases demonstrated similar activity to Bst polymerase without the Sso7d domain. At the same time, Btlv_Sso7d polymerase is characterized by increased reverse transcriptase activity and resistance to inhibitors, which allows it to be used in reagent kits for the diagnosis of infectious diseases by the LAMP method in combination with express nucleic acid extraction through thermal inactivation or lysis in the presence of guanidine chloride without subsequent washes from the components of the biological material and lysis solution. This significantly reduced the required time for the analysis.
As the demand for rapid and accurate diagnosis of infectious diseases grows, the use of LAMP methods, and consequently, Bst polymerase analogs with improved properties, will only increase.
About the authors
Tatyana L. Zamotaeva
Central Research Institute of Epidemiology
Email: sazonova@pcr.ms
ORCID iD: 0009-0003-9799-3749
Researcher, Center for development, product development and innovation
Russian Federation, MoscowEkaterina A. Dedyaeva
Central Research Institute of Epidemiology
Email: safronova.e@cmd.su
ORCID iD: 0000-0002-2501-0956
Technologist-developer, Center for development, product development and innovation
Russian Federation, MoscowOlga O. Mikheeva
Central Research Institute of Epidemiology
Email: olga.mikheeva.92@mail.ru
ORCID iD: 0000-0002-1721-5134
Researcher, Research group of genetic engineering and biotechnology, Department of molecular diagnostics and epidemiology
Russian Federation, MoscowMaria I. Pika
Central Research Institute of Epidemiology
Author for correspondence.
Email: m.zotova@cmd.su
ORCID iD: 0000-0002-3279-6811
Researcher, Research group of genetic engineering and biotechnology, Department of molecular diagnostics and epidemiology
Russian Federation, MoscowEvgeny A. Cherkashin
Central Research Institute of Epidemiology
Email: e.cherkashin@pcr.ms
ORCID iD: 0000-0002-3627-6047
Cand. Sci. (Chem.), Head, Center for development, product development and innovation
Russian Federation, MoscowAnna S. Cherkashina
Central Research Institute of Epidemiology
Email: cherkashina@pcr.ms
ORCID iD: 0000-0001-7970-7495
Cand. Sci. (Chem.), Head, Research group of genetic engineering and biotechnology
Russian Federation, MoscowVasily G. Akimkin
Central Research Institute of Epidemiology
Email: v.akimkin@cmd.su
ORCID iD: 0000-0003-4228-9044
Dr. Sci. (Med.), Professor, Academician of the Russian Academy of Sciences, Director
Russian Federation, MoscowReferences
- Рубель М.С., Дубровина И.А., Мясников В.А. и др. Сравнительная характеристика современных экспресс-методов диагностики инфекционных заболеваний, основанных на методе изотермической полимеразной цепной реакции. Вестник Российской Военно-медицинской академии. 2018;(1):160–3. Rubel M.S., Dubrovina I.A., Miasnikov V.A., et al. Comparative characteristics of modern express methods of diagnosing infectious diseases base on isothermal PCR technology. Bulletin of the Russian Military Medical Academy. 2018;(1):160–3. EDN: https://elibrary.ru/emrzyi
- Чемисова О.С., Цырулина О.А., Трухачев А.Л., Носков А.К. Сравнительный анализ методов изотермической амплификации нуклеиновых кислот. Журнал микробиологии, эпидемиологии и иммунобиологии. 2022;99(1):126–38. Chemisova O.S., Tsyrulina O.A., Trukhachev A.L., Noskov A.K. Comparative analysis of methods for isothermal amplification of nucleic acids. Journal Of Microbiology, Epidemiology and Immunobiology. 2022;99(1):126–38. DOI: https://doi.org/10.36233/0372-9311-176 EDN: https://elibrary.ru/qbqrwj
- Notomi T., Okayama H., Masubuchi H., et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):E63. DOI: https://doi.org/10.1093/nar/28.12.e63
- Смирнова Д.И., Петруша О.А., Грачёва А.В. и др. Быстрая диагностика генитального герпеса методом петлевой изотермической амплификации ДНК с флуоресцентной детекцией. Журнал микробиологии, эпидемиологии и иммунобиологии. 2019;96(6):40–6. Smirnova D.I., Petrusha O.A., Gracheva A.V., et al. Rapid diagnostics of genital herpes by loop-mediated isothermal amplification method with fluorescent detection. Journal of Microbiology, Epidemiology and Immunobiology. 2019;96(6):40–6. DOI: https://doi.org/10.36233/0372-9311-2019-6-40-46 EDN: https://elibrary.ru/yskkec
- Акимкин В.Г., Петров В.В., Красовитов К.В. и др. Молекулярные методы диагностики новой коронавирусной инфекции: сравнение петлевой изотермической амплификации и полимеразной цепной реакции. Вопросы вирусологии. 2021; 66(6):417–24. Akimkin V.G., Petrov V.V., Krasovitov K.V., et al. Molecular methods for diagnosing novel coronavirus infection: comparison of loop-mediated isothermal amplification and polymerase chain reaction. Problems of Virology. 2022;66(6): 417–24. DOI: https://doi.org/10.36233/0507-4088-86 EDN: https://elibrary.ru/bsgdlo
- Shirshikov F.V., Bespyatykh J.A. Loop-mediated isothermal amplification: from theory to practice. Russ. J. Bioorg. Chem. 2022;48(6):1159–74. DOI: https://doi.org/10.1134/S106816202206022X
- Пика М.И., Михеева О.О., Соловьева Е.Д. и др. Получение Bst-полимеразы для диагностики различных инфекций методом петлевой изотермической амплификации. Журнал микробиологии, эпидемиологии и иммунобиологии. 2023;100(3):210–8. Pika M.I., Mikheeva O.O., Solovyova E.D., et al. Production of Bst polymerase for diagnosis of different infections using loop-mediated isothermal amplification. Journal of Microbiology, Epidemiology and Immunobiology. 2023;100(3):210–8. DOI: https://doi.org/10.36233/0372-9311-364 EDN: https://elibrary.ru/phcmoq
- Wada K., Suzuki H. Biotechnological platforms of the moderate thermophiles, Geobacillus species: notable properties and genetic tools. In: Salwan R., Sharma V., eds. Physiological and Biotechnological Aspects of Extremophiles. Academic Press; 2020:195–218. DOI: https://doi.org/10.1016/C2018-0-03860-8
- Stenesh J., McGowan G.R. DNA polymerase from mesophilic and thermophilic bacteria. III. Lack of fidelity in the replication of synthetic polydeoxyribonucleotides by DNA polymerase from Bacillus licheniformis and Bacillus stearothermophilus. Biochim. Biophys. Acta. 1977;475(1):32–41. DOI: https://doi.org/10.1016/0005-2787(77)90336-7
- Chien A., Edgar D.B., Trela J.M. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J. Bacteriol. 1976;127(3):1550–7. DOI: https://doi.org/10.1128/jb.127.3.1550-1557.1976
- Oscorbin I., Filipenko M. Bst polymerase – a humble relative of Taq polymerase. Comput. Struct. Biotechnol. J. 2023;21:4519–35. DOI: https://doi.org/10.1016/j.csbj.2023.09.008
- Li P., Amenov A., Kalendar R., et al. Cloning and purification of large fragment of DNA polymerase I from geobacillus stearothermophilus and application in isothermal DNA amplification. Eurasian J. Appl. Biotechnol. 2017;(1):50–8. EDN: https://elibrary.ru/zbenmt
- Oscorbin I.P., Boyarskikh U.A., Filipenko M.L. Large fragment of DNA polymerase I from Geobacillus sp. 777: сloning and comparison with DNA polymerases I in practical applications. Mol. Biotechnol. 2015;57(10):947–59. DOI: https://doi.org/10.1007/s12033-015-9886-x
- Chander Y., Koelbl J., Puckett J., et al. A novel thermostable polymerase for RNA and DNA loop-mediated isothermal amplification (LAMP). Front. Microbiol. 2014;5:395. DOI: https://doi.org/10.3389/fmicb.2014.00395
- Wang Y., Prosen D.E., Mei L., et al. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res. 2004;32(3):1197–207. DOI: https://doi.org/10.1093/nar/gkh271
- Sidstedt M., Rådström P., Hedman J. PCR inhibition in qPCR, dPCR and MPS-mechanisms and solutions. Anal. Bioanal. Chem. 2020;412(9):2009–23. DOI: https://doi.org/10.1007/s00216-020-02490-2
- Oscorbin I.P., Belousova E.A., Boyarskikh U.A., et al. Derivatives of Bst-like Gss-polymerase with improved processivity and inhibitor tolerance. Nucleic Acids Res. 2017;45(16):9595–610. DOI: https://doi.org/10.1093/nar/gkx645
- Li J., Li Y., Li Y., et al. An enhanced activity and thermostability of chimeric Bst DNA polymerase for isothermal amplification applications. Appl. Microbiol. Biotechnol. 2023;107(21):6527–40. DOI: https://doi.org/10.1007/s00253-023-12751-6
- Yu Z., Wang J. Strategies and procedures to generate chimeric DNA polymerases for improved applications. Appl. Microbiol. Biotechnol. 2024;108(1):445. DOI: https://doi.org/10.1007/s00253-024-13276-2
- Paik I., Bhadra S., Ellington A.D. Charge engineering improves the performance of Bst DNA polymerase fusions. ACS Synth. Biol. 2022;11(4):1488–96. DOI: https://doi.org/10.1021/acssynbio.1c00559
- Ordóñez C.D., Lechuga A., Salas M., Redrejo-Rodríguez M. Engineered viral DNA polymerase with enhanced DNA amplification capacity: a proof-of-concept of isothermal amplification of damaged DNA. Sci. Rep. 2020;10(1):15046. DOI: https://doi.org/10.1038/s41598-020-71773-6
- Coulther T.A., Stern H.R., Beuning P.J. Engineering polymerases for new functions. Trends Biotechnol. 2019;37(10):1091–103. DOI: https://doi.org/10.1016/j.tibtech.2019.03.011
- Lischer K., Tansil K.P., Ginting M.J., et al. Cloning of DNA Polymerase I Geobacillus thermoleovorans SGAir0734 from a Batu Kuwung Hot Spring in Escherichia coli. Int. J. Technol. 2020;11(5):921–30. DOI: https://doi.org/10.14716/ijtech.v11i5.4311
- Gaultier N.E., Junqueira A.C.M., Uchida A., et al. Genome sequence of Geobacillus thermoleovorans SGAir0734, isolated from Singapore air. Genome Announc. 2018;6(27):e00636–18. DOI: https://doi.org/10.1128/genomea.00636-18
- Xiong A.S., Yao Q.H., Peng R.H., et al. A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res. 2004;32(12):e98. DOI: https://doi.org/10.1093/nar/gnh094
- Черкашина А.С., Михеева О.О., Пика М.И. и др. Способ получения большого фрагмента Bst-полимеразы (варианты). Патент РФ № 2 809 366;2023. Cherkashina A.S., Mikheeva O.O., Pika M.I., et al. Method for obtaining a large fragment of Bst polymerase (variants). Patent RF № 2 809 366;2023.
- Брагин А.Г., Глушков С.А., Иванов М.К. и др. Определение ДНК-полимеразной и нуклеазной активностей ДНК-зависимых полимераз с использованием флуоресцентной детекции в режиме реального времени. Биохимия. 2008;73(9)1252–64. EDN: https://elibrary.ru/jubdgz Bragin A.G., Glushkov S.A., Ivanov M.K., et al. Determination of DNA polymerase and nuclease activities of DNA-dependent polymerases using real-time fluorescent detection. Biochemistry. 2008;73(9):1007–17. DOI: https://doi.org/10.1134/S0006297908090083 EDN: https://elibrary.ru/lliwvl
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227(5259):680–5. DOI: https://doi.org/10.1038/227680a0
- Napoli A., Zivanovic Y., Bocs C., et al. DNA bending, compaction and negative supercoiling by the architectural protein Sso7d of Sulfolobus solfataricus. Nucleic Acids Res. 2002;30(12):2656–62. DOI: https://doi.org/10.1093/nar/gkf377
- Xiang R., Liu G.Y., Hou Y., et al. Double domain fusion improves the reverse transcriptase activity and inhibitor tolerance of Bst DNA polymerase. Int. J. Biol. Macromol. 2024;274(Pt. 1):133243. DOI: https://doi.org/10.1016/j.ijbiomac.2024.133243
- Hernández-Rollán C., Ehrmann A.K., Vlassis A., et al. Neq2X7: a multi-purpose and open-source fusion DNA polymerase for advanced DNA engineering and diagnostics PCR. BMC Biotechnol. 2024;24(1):17. DOI: https://doi.org/10.1186/s12896-024-00844-7
Supplementary files

