


Multilocus sequence-typing scheme for Borrelia miyamotoi — the erythema-free ixodid tick-borne borreliosis pathogens
- Authors: Mironov K.O.1, Titkov A.V.1, Kuleshov K.V.1, Platonov A.E.1
-
Affiliations:
- Central Research Institute for Epidemiology
- Issue: Vol 101, No 1 (2024)
- Pages: 80-88
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18555
- DOI: https://doi.org/10.36233/0372-9311-419
- EDN: https://elibrary.ru/wjphcn
- ID: 18555

Cite item

Abstract
Introduction. Borrelia miyamotoi is a pathogen of erythema-free ixodid tick-borne borreliosis (ITBB), a disease widespread in Russia. To date, there are no generally accepted methods for B. miyamotoi genotyping. The multilocus sequencing typing (MLST) scheme of Borrelia was originally developed for B. burgdorferi, and does not have the required discrimination power for monitoring the ITBB pathogens.
The objective of this study is to develop the MLST scheme for B. miyamotoi.
Materials and Methods. The whole genome sequences of 10 reference strains (GenBank) were analyzed for the selection of the house-keeping loci. The MLST scheme development was based on principles published by the authors of the method. For this experiment, 81 B. miyamotoi strains and positive clinical samples were used to test the MLST scheme.
Results. After analyzing the genomic data, 8 house-keeping loci were chosen for MLST, for which the PCR and sequencing primers were designed. Each MLST loci was represented by several alleles (from 4 to 7) which form 15 sequence types. The genetic diversity of pathogens isolated from ITBB patients and ticks were characterized.
Discussion. Based on pairwise distances between allelic profiles, the sequence types can be classified into four groups. The first two groups are clonal complexes; the other two groups are formed by once identified sequence types. The first clonal complex unites 11 sequence types (80 or 88% of the characterized B. miyamotoi), the second consists of 2 sequence types (9 or 9.8%). The genetic differences between B. miyamotoi are associated with the sources of strains and biological isolates. The MLST based classification confirms the previously described genetic heterogeneity of B. miyamotoi populations associated with ecologically unrelated vectors of ITBB pathogens.
Conclusion. The proposed MLST scheme is an appropriate tool for ITBB pathogen classification and evolutionary change characterization within clonal complexes.
Full Text
Introduction
Borrelia miyamotoi is the pathogen of the erythematous form of ixodid tick-borreliosis (ITBB) which belongs to the group of relapsing fever pathogens, but is transmitted by ticks of the Ixodes genus. The history of discovery, taxonomy, biological properties of pathogens, aspects of pathogenesis, clinic and epidemiology of ITBB were published in a monograph by A.E. Platonov in 2017 [1]. A relevant area of B. miyamotoi research is the development of intraspecies pathogen classification methods. This is due to the necessity to study the clinical features of ITBB caused by different genetic variants of the pathogen, including those associated with the phenomenon of immune evasion [1], as well as the necessity to solve classical epidemiological problems related to monitoring pathogens. Because of the severely limited use of mass parallel sequencing, which is primarily due to the difficulty of culturing B. miyamotoi, as well as the low concentration of pathogens in the biological material sample, there is a necessity to develop affordable polymerase chain reaction (PCR)-based and fragment sequencing-based methods for antigenic and genetic characterization of ITBB pathogens.
To characterize the antigenic diversity of B. miyamotoi, we have proposed a method of determining the main surface proteins [2, 3], which allows simultaneous detection of several antigenic variants and identification of the most clinically and epidemiologically significant variants of ITBB pathogens circulating in Russia. However, antigen detection cannot be used for the study of evolutionary processes occurring in the bacterial population to identify pathogens with increased virulent or pathogenic properties.
The triple-locus typing scheme based on the sequencing of p66, glpQ, and 16S gene fragments [4], proposed earlier by domestic researchers could be used to characterize representatives of the Borrelia genus, but this scheme does not have the necessary discrimination power for B. miyamotoi.
A convenient tool for studying the genetic properties of pathogens that are not under immune system pressure, allowing identification of individual strains and characterization of their genetic relationships, is the method of multilocus sequencing typing (MLST), which has been successfully used in epidemiological practice since 1998 [5, 6]. In addition to the previously demonstrated advantages of using multiple genetic loci in analyzing genetic relationships of pathogens, an important advantage of MLST is the absolute interlaboratory reproducibility of the results, which allows combining genetic and epidemiologic information into a single database [7]. Currently, the PubMLST Internet resource contains information on MLST schemes for more than 130 species of microorganisms; for many species, published MLST schemes are the golden standard for their intraspecific characterization. At the same time, the developed MLST scheme for bacteria of the Borrelia genus and its modifications do not have sufficient discrimination power to differentiate B. miyamotoi. This is due to the fact that the MLST scheme was initially developed for B. burgdorferi [8], which does not allow its use for monitoring Russian ITBB pathogens. Since the B. miyamotoi genome has significant differences from bacteria of the Borrelia burgdorferi sensu lato complex, additional analysis of available whole-genome sequences is necessary in order to develop and validate the B. miyamotoi MLST methodology.
Materials and methods
Nucleotide sequences
When selecting sequences for MLST, we used whole genome sequences of 6 Russian strains — Izh-4 (GenBank identification number CP024390.2, number in the State Collection of Pathogenic Microorganisms and Cell Cultures "SCPM-Obolensk" B-8324), Izh-5 (CP024205. 2, B-8325), Izh-14 (CP024371.2, B-8326), Izh-16 (CP024351.2, B-8327), Yekat-1 (CP024333.2, B-8328), Yekat-6 (CP024316.2, B-8329) [1, 9] as well as 4 foreign strains – FR64b (CP004217. 2), HT-31 (CP114703.1), LB-2001 (CP114690.1) and CA17-2241 (CP021872.1) [1, 10, 11], known since the beginning of the study.
Strains and samples of biological material
During development, the MLST scheme was tested on 7 strains, NL-IR-1 (CP044783.1), Yekat-18 (CP037471.1, B-8810), Yekat-76 (CP037058.1, B-8814), Yekat-19 (CP036557.1, B-8811), Yekat-17 (CP037215. 2, B-8330), Yekat-21 (CP036914.2, B-8812), Yekat-31 (CP036726.1, B-8813) and 81 B. miyamotoi DNA samples isolated in 2012-2022 from 50 tick suspensions and the blood of 31 ITBB patients. The suspensions were obtained from Ixodes vector ticks collected in Western Europe and European regions of Russia (I. ricinus), as well as central and eastern regions of Russia (I. persulcatus). Most of the biological samples were obtained and characterized during the development of a methodology for determining the B. miyamotoi main surface protein antigenic properties [2]. The study was conducted with voluntary informed consent of patients. The study protocol was approved by the Ethical Committee of the Central Research Institute of Epidemiology of Rospotrebnadzor (protocol No. 83 of 26.06.2018).
Furthermore, 9 strains whose whole-genome sequences were used to select MLST loci were studied. The characteristics of the studied strains and biological material samples are given in Table 1.
Table 1. The B. miyamotoi DNA samples sources and their genetic characteristics (sequence types and clonal complex)
DNA source | Sequence type | Сlonal complex | Territory | Years | Number of samples |
Blood of patients with ixodid tick-borne borreliosis caused by B. miyamotoi (n = 37) | ST-1* | I | Sverdlovsk Region Udmurt Republic | 2016–2018 2016 | 19 1 |
ST-2* | I | Sverdlovsk Region Udmurt Republic | 2017 2016 | 2 1 | |
ST-3 | I | Udmurt Republic | 2016 | 1 | |
ST-4 | I | Sverdlovsk Region | 2016 | 1 | |
ST-5* | I | Udmurt Republic | 2016 | 1 | |
ST-12* | I | Sverdlovsk Region Krasnoyarsk Territory | 2017 2017 | 1 1 | |
ST-13 | I | Sverdlovsk region | 2017 | 1 | |
ST-14* | I | Novosibirsk Region Khabarovsk Territory Sverdlovsk Region Krasnoyarsk Territory | 2012 2012 2017 2017, 2019 | 1 2 2 3 | |
Suspensions of ticks of Ixodes genus (n = 54) | ST-1* | I | Novosibirsk Region Sverdlovsk Region Samara Region Omsk Region | 2014, 2017 2013–2014 2019 2022 | 4 3 1 5 |
ST-2* | I | Altai Republic Samara Region | 2016 2019 | 1 1 | |
ST-5* | I | Omsk Region | 2022 | 1 | |
ST-6 | I | Japan | 1992 | 1 | |
ST-7 | I | Japan | 1990 | 1 | |
ST-8 | – | Connecticut, USA | 2001 | 1 | |
ST-9 | – | California, USA | 2015 | 1 | |
ST-10 | II | Netherlands Czech Republic Kabardino-Balkarian Republic | 2018 2019 2021 | 1 6 1 | |
ST-11 | II | Kabardino-Balkarian Republic | 2021 | 1 | |
ST-12* | I | Novosibirsk Region Omsk Region | 2017 2022 | 4 3 | |
ST-14* | I | Altai Republic Amur Region Novosibirsk Region Omsk Region | 2016 2016 2017 2022 | 6 1 9 1 | |
ST-15 | I | Tomsk Region | 2014 | 1 | |
Note. *Sequence types found both in patient samples and ticks. | |||||
PCR and sequencing
DNA extraction and PCR were performed using reagents produced by the Central Research Institute of Epidemiology of Rospotrebnadzor. Amplification and sequencing of gene fragments were performed with primers (Table 2) according to the following procedure: 95ºC — 15 min (1 cycle), 95ºC — 10 s, 60ºC — 20 s, 72ºC — 20 s (40 cycles). The PCR and sequencing methods carried out with the help of Applied Biosystems equipment and reagents were similar to those used in previous studies [2, 12].
Table 2. Fragments of genes for MLST, PCR and sequencing primers
Fragment name | Protein* | 5’-3’ primer sequences | The length of the PCR products, bp | The length of the MLST loci, bp |
acpS | holo-ACP synthase | gACgAAATCAATAggATgTgATATAATAAAggT CTATTACAAATgCAATAgAgTACTCCCTTTCA | 365 | 192 |
nusB | Transcription antitermination factor NusB | ggATTTAAgACATAAggCTAgAgTTTTAgCTTTTC gAgCTCTCCATATTTTTTAACAAAgCATCAAg | 417 | 380 |
motB | Flagellar motor protein MotB | CCTgAATATATgTTgACATATggAgACATggTT CCTgCAAATCCAgATACCTCAAATTTACTC | 623 | 413 |
dnaX | DNA polymerase III subunit gamma/tau | CTgCTATTAAgAAgCgTCCCAgAgAT CTgATCAAAAAgAgTATAAgCATCCCTTACAC | 653 | 560 |
rplP | 50S ribosomal protein L16 | gTATAgAAAgAAgCAAAgAggAAgACTgTCA CACCTCAAATCTCgCCTTATCACAAATATAg | 393 | 269 |
сdd | Cytidine deaminase | GAAGCTGCAAGAAATAATGCATATTCACCAT GCTGCATAATCCTATTATTTGTAAAGATAGC | 620 | 233 |
lysM | LysM peptidoglycan-binding domain-containing protein | gACTTgATCCTggTgCTATTgTTAAAgCTAg CCCgATCTCTAAgCTTAgAAgATCTAATTgC | 624 | 522 |
miaA | tRNA (adenosine(37)-N6)-dimethylallyltransferase MiaA | TCCTACgggTgTAggTAAAAgTgACATT CCTCTTCAAgCAATCCACAATCAATC | 626 | 555 |
Note. *The designation of the protein from NCBI (www.ncbi.nlm.nih.gov). **The nucleotide positions of MLST loci in the reference strains were presented in the Table 3.
Analysis of MLST data
Sequencing results with allele designations and sequence types (ST) were processed in accordance with the method developer requirements [5–7]. ST analysis using BURST and UPGMA algorithms and genetic tree construction were carried out using the "START2 v. 1.0.5" software [13].
Results
In accordance with the MLST principles, the selection of DNA fragments for allele designation (MLST loci) was subject to the following conditions: the fragments should be located in unlinked genes within the chromosome and represented by several alleles; furthermore, the combinations of alleles should allow for intraspecies pathogen classification [5, 6]. Whole-genome sequence analysis of 6 strains allowed us to identify several dozen groups of unlinked genetic loci, 8 of which were sequentially selected (Table 2), allowing us to differentiate epidemiologically unrelated strains which were isolated at different times, in different territories, and from different sources. Table 3 presents our proposed allele designations and profiles that determine ST strains and B. miyamotoi DNA in biological samples. For allele designations, the MLST loci coordinates in the whole-genome nucleotide sequences of the reference strains are given, except for the nusB-6 and lysM-7 alleles, for which identification numbers in the GenBank database are given. Differences in the MLST loci are represented by single-nucleotide substitutions, except for the nusB fragment, for which 2 alleles (nusB-4 and nusB-6 in ST-10 and ST-11 allele profiles) containing a 6-nucleotide insertion were identified.
Table 3. Designations of alleles and sequence types
DNA source | Fragments (lenght, bp)*** | Sequence type | ||||||||
acpS (192) | nusB (380, 386) | motB (413) | dnaX (560) | rplP (269) | сdd (233) | lysM (522) | miaA (555) | |||
Strains** | Yekat-1* | 1 896361–896170 | 1 800974–801353 | 1 613232–613644 | 1 423437–423996 | 1 404829–405097 | 1 265228–265460 | 1 253627–254148 | 1 37083–37637 | ST-1 |
Izh-14 | 1 | 1 | 1 | 1 | 2 404724–404992 | 1 | 1 | 1 | ST-2 | |
Izh-4 | 1 | 1 | 1 | 1 | 1 | 1 | 2 253612–254133 | 2 37068–37622 | ST-3 | |
Yekat-6 | 1 | 2 800953–801332 | 2 613211–613623 | 1 | 1 | 1 | 1 | 1 | ST-4 | |
Izh-5 | 2 896194–896385 | 1 | 1 | 2 423437–423996 | 1 | 2 265228–265460 | 1 | 1 | ST-5 | |
FR64b | 2 | 1 | 1 | 2 | 1 | 1 | 3 650921–651442 | 1 | ST-6 | |
HT31 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 3 37098–37652 | ST-7 | |
LB-2001 | 3 897013–897204 | 3 801812–802191 | 3 613265–613677 | 3 423552–424111 | 3 404960–405228 | 3 265566–265798 | 4 253956–254477 | 4 37153–37707 | ST-8 | |
CA17-2241 | 5 10054–10245 | 5 105075–105454 | 5 292933–293345 | 6 482530–483089 | 5 501393–501661 | 5 640132–640364 | 6 651447–651968 | 6 868175–868729 | ST-9 | |
NL-IR-1 | 4 893710–893901 | 4 798494–798879 | 4 611883–612295 | 4 422636–423195 | 4 404082–404350 | 4 265475–265707 | 5 253875–254396 | 5 37104–37658 | ST-10 | |
DNA samples | 136_KBR21 | 4 | 6 | 4 | 4 | 4 | 4 | 5 | 5 | ST-11 |
Bal_Y17 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ST-12 | |
Sha_Y17 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | ST-13 | |
4426_Y17 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | ST-14 | |
2154_T14 | 2 | 1 | 1 | 1 | 1 | 1 | 7 | 1 | ST-15 | |
Note. *The Izh-16 strain is assigned to ST-1. **Alleles and sequence types of the first nine strains were designated based on the whole genome sequences, allelic profiles of the NL-IR-1 strain and isolates were obtained by testing the MLST scheme. ***For the unique alleles the nucleotide positions of MLST loci in the reference strains sequences described in Material and Methods section are indicated. The alleles nusB-6 and lysM-7 have identification numbers in the GenBank database OR192576 and OR134830, respectively.
When developing the in silico MLST scheme, 9 STs were found in 10 strains. Sequencing of gene fragments included in the MLST scheme was performed for 9 strains, and no discordant results were found in comparison with the data of whole-genome sequencing presented in GenBank. Further analysis of nucleotide sequences from DNA samples revealed 6 more STs, of which 2 were formed by two new alleles and 4 by new combinations of previously identified alleles. ST-1 (33; 36%) and ST-14 (25; 27.4%) were the most frequent in the studied sample, ST-12 (9; 10%), ST-10 (8; 9%), ST-2 (5; 5.5%) and ST-5 (2; 2.2%) were less frequent. ST-3, ST-4, ST-6, ST-7, ST-8, ST-9, ST-11, ST-13 and ST-15 occurred once (1.1% each).
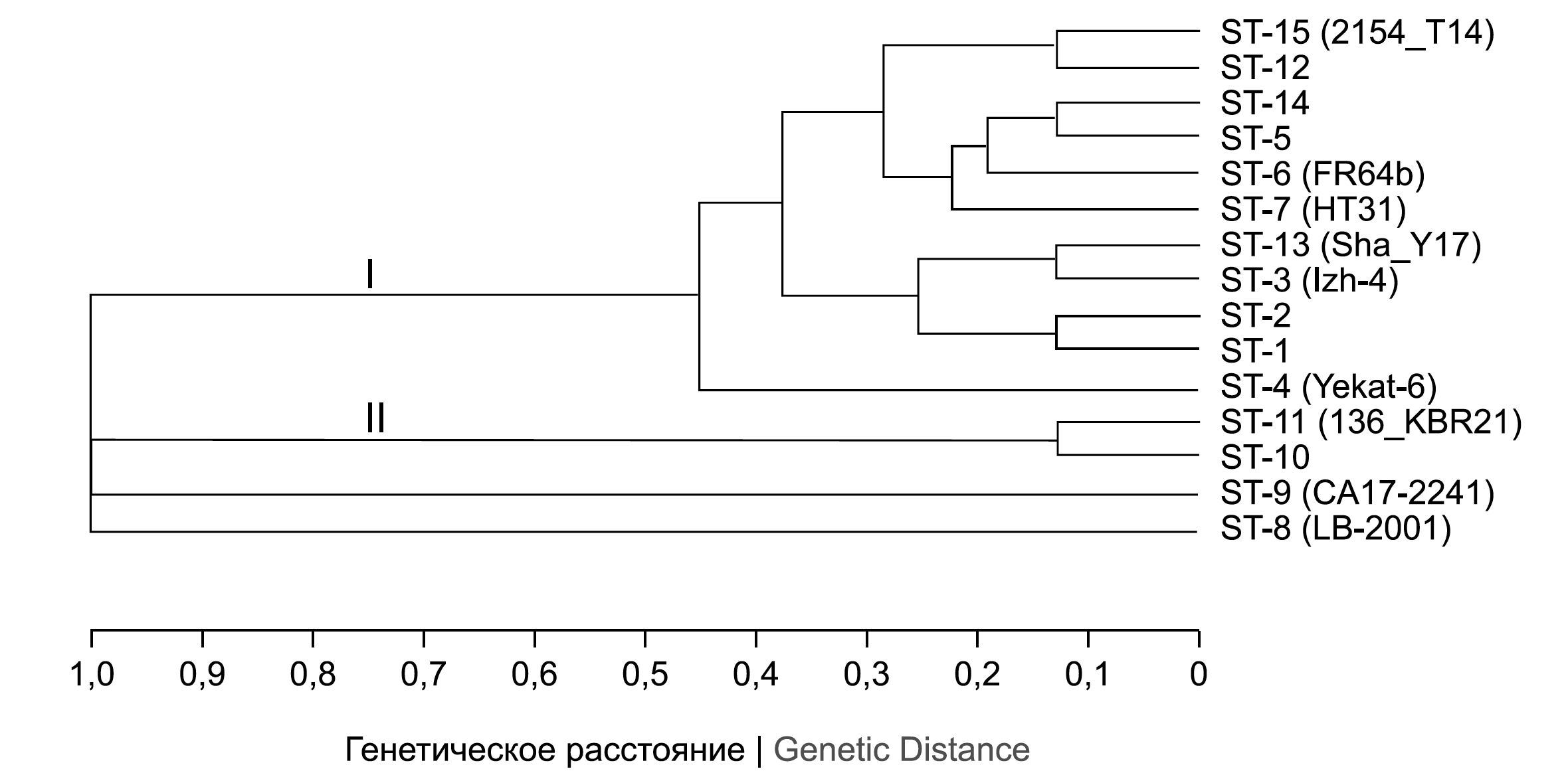
The figure presents a dendrogram illustrating the genetic relationships of ST. The dendrogram and the results presented in Table 1 combine data on 91 representatives of the B. miyamotoi species from different sources.

Genetic relationships of sequence types within the B. miyamotoi species based on allelic profiles differences (the UPGMA algorithm). The number in parentheses is the strain or isolates name for the sequence types found once. The Roman numeral is the designation of clonal complexes.
Discussion
The genetic relationships of B. miyamotoi presented in the figure, the determination of which was based on the analysis of the number of mismatches in allelic profiles, allow us to classify the designated STs into 4 groups. The first 2 groups correspond to the clonal complexes labeled I and II in the figure, the other 2 groups are formed by ST-8 and ST-9, which have differences in all 8 loci from each other and from the other STs.
The first clonal complex (I) brings together 11 STs which are found in 80 (88%) of the characterized strains and DNA samples. ST-14 can be designated as the central one, because the allelic profile corresponding to it has the maximum number of mismatches at 1 MLST locus from the allelic profiles of other STs of this clonal complex, equal to 4. The second clonal complex (II) is formed by ST-10 and ST-11 which are found in 8 (8.7%) DNA samples and 1 (1.1%) strain. The allelic profiles of ST-10 and ST-11 have mismatches at 1 MLST locus and differ from the allelic profiles of the first clonal complex STs at all 8 MLST loci.
Table 1 presents STs and characterization of B. miyamotoi DNA sources. Most of the characterized DNA samples have STs found both in samples from ITBB patients and in tick suspensions. ST-3, ST-4, and ST-13 found once in ITBB pathogens and ST-6, ST-7, ST-11, and ST-15 found in tick suspensions do not allow the determination of specific genotypes associated with ITBB cases. Presumably, all representatives of the first clonal complex found in vectors can cause ITBB.
The pronounced genetic differences of characterized B. miyamotoi are related to the B. miyamotoi DNA sources. All the pathogens included in the first clonal complex were isolated from I. persulcatus tick suspensions obtained in Russia. The first clonal complex also contains strains FR64b and HT31 isolated in Japan from the same vector species. STs included in the second clonal complex were found in I. ricinus tick suspensions obtained from European countries and in the Kabardino-Balkar Republic. ST-8 and ST-9 B. miyamotoi were isolated from ticks of I. scapularis and I. pacificus species, respectively, distributed in North America.
Thus, the proposed classification based on MLST confirms the previously demonstrated genetic heterogeneity of B. miyamotoi populations corresponding to Asian (clonal complex I), European (clonal complex II) and American (ST not included in clonal complexes I and II) genotypes [1, 14, 15], circulating in different continents, which is associated with ecologically unrelated vectors of the pathogens.
Conclusion
In this study, based on the available whole-genome data, an MLST scheme was developed to differentiate B. miyamotoi pathogens collected in different territories. By the example of the first ST clonal complex distribution, it is possible to draw a conclusion about the most common variants of B. miyamotoi circulating in Russia and some foreign countries. This approach may become a convenient tool for monitoring the pathogens of ITBB and characterizing evolutionary changes in the described clonal complexes. The use of MLST involves the examination of biological material samples after qualitative detection of B. miyamotoi DNA or obtaining a culture of the pathogen. Due to the complexity of B. miyamotoi cultivation, the bulk of B. miyamotoi pathogens can be characterized using MLST and the rest are characterized with the help of whole-genome sequencing, which at the present stage is necessary for a thorough analysis of evolutionary changes occurring in bacterial populations.
Further studies should be focused on expanding the selection of characterized isolates, including validation of the developed in silico MLST scheme using nucleotide sequence information published in GenBank, as well as searching for possible relationships between the described B. miyamotoi ST and antigenic variants determined with the help of the main surface protein typing data [2]. After extended testing of the proposed MLST scheme, the possibility of combining all the obtained data into a common database, similar to existing standards for intraspecific characterization of other pathogens, may be considered [7].
About the authors
Konstantin O. Mironov
Central Research Institute for Epidemiology
Author for correspondence.
Email: mironov@pcr.ru
ORCID iD: 0000-0001-8207-9215
D. Sci. (Med.), Head, Laboratory of molecular methods for genetic polymorphisms research, Central Research Institute for Epidemiology
Russian Federation, MoscowAnton V. Titkov
Central Research Institute for Epidemiology
Email: mironov@pcr.ru
ORCID iD: 0000-0001-7548-9267
researcher, Laboratory of zoonoses, Central Research Institute for Epidemiology
Russian Federation, MoscowKonstantin V. Kuleshov
Central Research Institute for Epidemiology
Email: mironov@pcr.ru
ORCID iD: 0000-0002-5238-7900
Cand. Sci. (Biol.), Head, Laboratory of molecular diagnostics and epidemiology of intestinal infections, Central Research Institute for Epidemiology
Russian Federation, MoscowAlexander E. Platonov
Central Research Institute for Epidemiology
Email: mironov@pcr.ru
ORCID iD: 0000-0001-7450-0081
D. Sci. (Biol.), Prof., chief researcher, Laboratory of zoonoses, Central Research Institute for Epidemiology
Russian Federation, MoscowReferences
- Платонов А.Е. «Новая» инфекция, вызываемая Borrelia miyamotoi: микробиология, эпидемиология, диагностика, клиника и патогенез. М.;2017. Platonov A.E. A «New» Infection Caused by Borrelia miyamotoi: Microbiology, Epidemiology, Diagnostics, Clinic and Pathogenesis. Moscow;2017. EDN: https://elibrary.ru/yabrqr
- Миронов К.О., Титков А.В., Кулешов К.В. и др. Разработка и практическое применение методики для идентификации поверхностных антигенов Borrelia miyamotoi. Журнал микробиологии, эпидемиологии и иммунобиологии. 2021;98(3):339–50. Mironov K.O., Titkov A.V., Kuleshov K.V., et al. Development and application of the technique for identification of Borrelia miyamotoi surface antigens. Journal of Microbiology, Epidemiology and Immunobiology. 2021;98(3):339–50. DOI: https://doi.org/10.36233/0372-9311-142 EDN: https://elibrary.ru/kmeswh
- Миронов К.О., Титков А.В., Платонов А.Е. Комплекс молекулярно-биологических методик для внутривидовой характеристики бактерий вида Borrelia miyamotoi. Национальные приоритеты России. 2021;42(3):208–11. Mironov K.O., Titkov A.V., Platonov A.E. Complex of molecular biological techniques for Borrelia miyamotoi typing. National Priorities of Russia. 2021;42(3):208–11. EDN: https://elibrary.ru/kmeswh
- Фоменко Н.В., Боргояков В.Ю., Панов В.В. Генетические особенности ДНК боррелий вида Borrelia miyamotoi, выявляемых в таежных клещах. Молекулярная генетика, микробиология и вирусология. 2011;(2):12–7. Fomenko N.V., Borgoyakov V.Yu., Panov V.V. Genetic features of DNA of Borrelia miyamotoi transmitted by Ixodes persulcatus. Molecular Genetics, Microbiology and Virology. 2011;(2):12–7. EDN: https://elibrary.ru/nwewlr
- Maiden M.C., Bygraves J.A., Feil E., et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl Acad. Sci. USA. 1998;95(6):3140–5. DOI: https://doi.org/10.1073/pnas.95.6.3140
- Платонов А.Е., Шипулин Г.А., Платонова О.В. Мультилокусное секвенирование – новый метод генотипирования бактерий и первые результаты его применения. Генетика. 2000;36(5):597–605. Platonov A.E., Shipulin G.A., Platonova O.V. Multilocus sequence typing: a new method and the first results in the genotyping of bacteria. Genetika. 2000;36(5):597–605. EDN: https://elibrary.ru/mpfxxb
- Jolley K.A., Bray J.E., Maiden M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124. DOI: https://doi.org/10.12688/wellcomeopenres.14826.1
- Margos G., Gatewood A.G., Aanensen D.M., et al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl Acad. Sci. USA. 2008;105(25):8730–5. DOI: https://doi.org/10.1073/pnas.0800323105
- Kuleshov K.V., Koetsveld J., Goptar I.A., et al. Whole-genome sequencing of six Borrelia miyamotoi clinical strains isolated in Russia. Genome Announc. 2018;6(1):e01424-17. DOI: https://doi.org/10.1128/genomeA.01424-17
- Fukunaga M., Takahashi Y., Tsuruta Y., et al. Genetic and phenotypic analysis of Borrelia miyamotoi sp. nov., isolated from the ixodid tick Ixodes persulcatus, the vector for Lyme disease in Japan. Int. J. Syst. Bacteriol. 1995;45(4):804–10. DOI: https://doi.org/10.1099/00207713-45-4-804
- Kingry L.C., Replogle A., Dolan M., et al. Chromosome and large linear plasmid sequences of a Borrelia miyamotoi strain isolated from Ixodes pacificus ticks from California. Genome Announc. 2017;5(37):e00960-17. DOI: https://doi.org/10.1128/genomeA.00960-17
- Миронов К.О., Платонов А.Е., Королева И.С., Шипулин Г.А. Анализ московской популяции штаммов Neisseria meningitidis методом мультилокусного секвенирования-типирования. Журнал микробиологии, эпидемиологии и иммунобиологии. 2006;93(2):31–6. Mironov K.O., Platonov A.E., Koroleva I.S., Shipulin G.A. Analysis of the Moscow population of Neisseria meningitidis strains by the method of multilocus sequencing-typing. Journal of Microbiology, Epidemiology and Immunobiology. 2006;93(2):31–6. EDN: https://elibrary.ru/htqatx
- Jolley K.A., Feil E.J., Chan M.S., Maiden M.C. Sequence type analysis and recombinational tests (START). Bioinformatics. 2001;17(12):1230–1. DOI: https://doi.org/10.1093/bioinformatics/17.12.1230
- Platonov A.E., Karan L.S., Kolyasnikova N.M., et al. Humans infected with relapsing fever spirochete Borrelia miyamotoi, Russia. Emerg. Infect. Dis. 2011;17(10):1816–23. DOI: https://doi.org/10.3201/eid1710.101474 EDN: https://elibrary.ru/pbdedx
- Crowder C.D., Carolan H.E., Rounds M.A., et al. Prevalence of Borrelia miyamotoi in Ixodes ticks in Europe and the United States. Emerg. Infect. Dis. 2014;20(10):1678–82. DOI: https://doi.org/10.3201/eid2010.131583
Supplementary files

