


Genetic characteristics of influenza A and B viruses circulating in Russia in 2019–2023
- Authors: Yatsyshina S.B.1, Artamonova A.A.1, Elkina M.A.1, Valdokhina A.V.1, Bulanenko V.P.1, Berseneva A.A.1, Akimkin V.G.1
-
Affiliations:
- Central Research Institute for Epidemiology
- Issue: Vol 101, No 6 (2024)
- Pages: 719-734
- Section: ORIGINAL RESEARCHES
- URL: https://microbiol.crie.ru/jour/article/view/18510
- DOI: https://doi.org/10.36233/0372-9311-480
- EDN: https://elibrary.ru/knbqnk
- ID: 18510

Cite item

Abstract
Relevance. Influenza viruses have a high potential for genetic change. These viruses are monitored annually around the world, including Russia, to determine the dominant genetic groups and select the strains to be included in influenza vaccines.
Objectives of the study include: analysis of influenza virus circulation in Russia in 2019–2023, phylogenetic and molecular analysis of hemagglutinin (HA) sequences of influenza viruses, detection of mutations associated with drug resistance to neuraminidase (NA) inhibitors and M2-protein (M2) ion channel inhibitors.
Materials and methods. Biological samples containing RNA of influenza viruses were studied: 410 A(H1N1)pdm09, 147 A(H3N2) and 167 B(Victoria). Sequencing of the HA, NA, M fragments was performed on the 3500xL Genetic Analyzer (Applied Biosytems). Data processing and analysis were carried out using DNASTAR, Nextclade, FluSurver and BioNumerics v.6.6 software.
Results. Influenza A(H1N1)pdm09, A(H3N2), B(Victoria) viruses circulating in 2019-2023 were investigated. The highest variability of HA was observed in A(H3N2) viruses. All influenza A(H1N1)pdm09 viruses in the 2022–2023 season had a previously unknown mutation E224A in HA, which increases its affinity for α-2,3 sialic acids — receptors localized in the human lungs, to which the virus binds via HA. 2 and 3% of influenza A(H1N1)pdm09 viruses in 2019–2020 and 2022–2023, respectively, had the D222N mutation in the receptor-binding site of HA, which is associated with more severe disease. The oseltamivir and zanamivir resistance mutation H275Y in NA was detected in 2.3% of influenza A(H1N1)pdm09 viruses in 2022–2023. No oseltamivir and zanamivir resistance mutations in NA were detected in all tested influenza A(H3N2) and B viruses. Sequencing data revealed a mutation of adamantane resistance S31N in M2 in all studied influenza viruses A(H1N1)pdm09 and A(H3N2).
Conclusions. The detection of amino acid substitutions in HA antigenic sites and resistance mutations in NA and M2 confirms the evolution of influenza viruses and the necessity for continuous genetic surveillance. The vast majority of currently circulating viruses remain sensitive to NA inhibitors.
Full Text
Introduction
Influenza viruses are enveloped viruses of the Orthomyxoviridae family, which are classified into 4 genera: influenza A, B, C and D viruses. Influenza A and B viruses pose the most significant danger to human health [1].
Influenza A viruses are the cause of most annual epidemics and all recurrent human pandemic diseases. They are subdivided into subtypes according to the combination of 2 surface glycoproteins located in the lipid membrane of virions: hemagglutinin (HA) and neuraminidase (NA). In birds, the main natural reservoir of influenza A viruses, 16 HA and 9 NA have been described; another 2 HA and NA have been found in bats [2]. Currently, influenza A viruses of the H1N1pdm09 and H3N2 subtypes cause the greatest number of epidemic diseases in humans1.
Influenza B viruses are specific to humans (cases in seals have also been described [3]) and are divided into 2 evolutionary lineages with significantly different antigenic properties (B/Victoria and B/Yamagata).
Influenza viruses are in a state of continuous evolution in their reservoirs, facilitated by a high mutation rate due to the absence of the proofreading activity of RNA-dependent RNA polymerase [4]. The main reservoir for influenza A viruses is wild migratory birds and other mammals, including humans, while influenza B and C viruses have no reservoir in the wild. Cumulative changes in sequences encoding HA and NA lead to antigenic drift of influenza A and B viruses: the structure of antigenic surfaces recognized by specific antibodies changes, contributing to the annual epidemic [5]. Antigenic shift is also possible for influenza A viruses: segments encoding HA (and to a lesser extent NA) that have evolved in animal influenza viruses can combine with human influenza virus segments to form new reassortant strains capable of causing pandemics [4, 5]. Since 1889, there have been 5 known pandemics of influenza A viruses, the most serious of which in 1918 was caused by the H1N1 subtype and the most recent in 2009 by the A(H1N1)pdm09 subtype [4, 6]. The H3N2 subtype began circulating in the 1968 pandemic [7]. Since 1971, both subtypes of influenza A virus — H3N2 and H1N1 (since 2009 — H1N1pdm09) and both antigenic lineages of influenza B virus have circulated annually with greater or lesser intensity.
Influenza B virus was identified in 1940.Two antigenic virus lineages, B/Victoria/2/87-like (Victoria) and B/Yamagata/16/88-like (Yamagata), have been co-circulating since 1983 [8].
B/Victoria influenza viruses evolve more rapidly with greater positive selection pressure than B/Yamagata lineage viruses [9], and the latter have not circulated since March 2020: since then, there have been sporadic reports of B/Yamagata influenza viruses, but without confirmation by HA sequencing of the virus2.
Under the auspices of WHO, influenza viruses are monitored worldwide, including their typing, genetic characterization and antigenic properties, to select among the predominant antigenic groups of virus strains to be included in influenza vaccines in the next epidemic season. The selection of vaccine strains with specific immunogenic properties is necessary to ensure an immune response against influenza viruses [10]. Large-scale genetic monitoring allows deep differentiation into clades and subclades and defines the trend of influenza virus evolution: the emergence, spread and disappearance from circulation of certain genetic variants.
During evolution, the most significant changes affect the HA of influenza viruses. The HA glycoprotein of influenza viruses is synthesized as a single polypeptide chain, which is further proteolytically cleaved into 2 subunits: HA1 and HA2. HA1 is responsible for binding the virus to sialic acids (SAs) on the cell membrane surface, while HA2 ensures fusion of the virus and endosome membranes.
Antigenic sites located at the HA1 apex near the receptor-binding site are the main targets for human neutralizing antibodies. HA glycosylation is associated with many properties, including immunogenicity and receptor specificity, and plays an important role in protecting antigenic sites from neutralizing antibodies [11]. The glycosylation pattern is more variable in HA1 than in HA2, which is more conserved [12].
Amino acid substitutions in the receptor-binding domain of HA affect the ability to bind to the host cell surface, which alters the virulence of the virus [13]. On the surface of human upper respiratory tract cells, predominantly SA-α2,6 are located, whereas in the lower respiratory tract, SA-α2,3 are located. Increased affinity of the virus for α-2,3-SAs may increase the severity of the disease [14].
The evolution of influenza B/Victoria viruses, in addition to point mutations, is facilitated by insertions and deletions of amino acids in the HA receptor binding region, which leads to immune evasion [15].
Amino acid substitutions in NA (influenza A and B viruses) and M2 (influenza A viruses) reduce the efficacy of drugs: NA inhibitors (oseltamivir, zanamivir) and adamantanes (amantadine and rimantadine), inhibitors of the M2 ion channel. According to the U.S. Centers for Disease Control and Prevention, virtually 100% of influenza A viruses are currently resistant to amantadine and rimantadine [16].
The objectives of this study are the molecular genetic analysis of HA, NA and M segments of influenza A(H1N1)pdm09, A(H3N2) and B/Victoria viruses circulating in Russia during the epidemic seasons 2019–2023, identification of amino acid substitutions in HA compared to vaccine strains, their possible impact on antigenic properties and strength of binding to specific receptors; phylogenetic analysis on the HA gene sequences, as well as analysis of NA and M2 for the presence of molecular markers of resistance to antiviral drugs.
Materials and methods
Detection of influenza A and B virus RNA in biological material (nasopharyngeal and oropharyngeal swabs, sputum, tracheal aspirates, bronchoalveolar lavage) obtained as a result of routine monitoring for influenza viruses was carried out in laboratories of the Hygiene and Epidemiology Centers of Rospotrebnadzor in 50 regions of the Russian Federation (Central, Northwestern, Southern, North Caucasus, Volga, Urals, and Far Eastern federal districts) by polymerase chain reaction (PCR) with hybridization-fluorescence detection of amplification products. The study was conducted with the informed consent of the patients. The research protocol was approved by the Ethics Committee of the Central Research Institute for Epidemiology (protocol No. 3, March 27, 2020). In case of unfavorable outcome of the disease, postmortem material (lung autopsy specimens) was examined.
For molecular genetic analysis by nucleic acid sequencing, biological material was submitted to the Reference Center for monitoring of upper and lower respiratory tract infections at the Laboratory of Molecular Diagnostics and Epidemiology of Respiratory Tract Infections of the Central Research Institute of Epidemiology.
Extraction of influenza virus RNA from biological material and the subsequent reverse transcription reaction were performed using the RIBO-prep and REVERTA-L reagent kits (AmpliSens). PCR for influenza virus RNA detection was performed using AmpliSens Influenza virus A/B, AmpliSens Influenza virus A-type-FL, AmpliSens Influenza virus A/H1-swine-FL and AmpliSens Influenza virus B-type-FL reagent kits (produced by the Central Research Institute of Epidemiology).
To amplify fragments of HA, NA, M genes of influenza viruses A/H1N1pdm09 and A/H3N2, HA and NA genes of influenza B viruses, PCR with electrophoresis detection was performed on thermocyclers (DNA-Technology) using AmpliSens reagents (Central Research Institute of Epidemiology).
Amplified fragments of individual segments of influenza viruses (NA, NA, M) were sequenced in the Scientific Group of Genetic Engineering and Biotechnology of the Central Research Institute of Epidemiology by the Sanger method using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems by Thermo Fisher Scientific) on the 3500xL Genetic Analyzer sequencer (Applied Biosystems).
Sequencing results were analyzed in the Reference Center for monitoring of upper and lower respiratory tract infections using the DNASTAR software block (SeqMan, EditSeq, MegAlign). The obtained nucleotide sequences were uploaded to the international database GISAID, can be filtered by CRIE Search patterns. Phylogenetic analysis was performed using the BioNumerics v. 6.6 program with the UPGMA method. Nextclade, FluSurver online platforms were used to track amino acid mutations. Amino acid numbering is given according to the corresponding influenza virus subtype.
Results
Biological samples containing RNA of influenza viruses: 410 A(H1N1)pdm09, 147 A(H3N2), and 167 B(Victoria) circulating in the epidemic seasons 2019–2020, 2020–2021, 2021–2022, 2022–2023 were analyzed. Influenza A(H1N1)pdm09 and influenza B virus were both detected in 1 sample (upper respiratory swabs) from the 2022–2023 season. Figure 1 shows that the number of influenza viruses tested varied in different years, which is due to the different intensity of the influenza epidemic process in different seasons.

Fig. 1. Number of influenza viruses tested at the Reference center for monitoring upper and lower respiratory tract infections in 2019–2023.
The nucleotide sequences of NA, HA, M genes of influenza A(H1N1)pdm09 and A(H3N2) viruses, and NA and HA genes of influenza B virus were obtained and analyzed (Table 1).
Table 1. Number of sequenced segments
Influenza virus | Segment | Number (from the recovered/from the deceased) | |||
2019–2020 | 2020–2021 | 2021–2022 | 2022–2023 | ||
(H1N1)pdm09 | HA | 59 (55/4) | – | – | 351 (296/55) |
NA | 59 (55/4) | – | – | 87 (69/18) | |
M | 59 (55/4) | – | – | 28 (16/2) | |
A(H3N2) | HA | 11 (11/0) | 2 (1/0) | 124 (124/0) | 10 (10/0) |
NA | 2 (2/0) | 1 (1/0) | 44 (44/0) | 1 (1/0) | |
M | 1 (1/0) | 2 (1/0) | 34 (34/0) | 1 (1/0) | |
В | HA | 56 (53/3) | – | 7 (7/0) | 104 (102/2) |
NA | 35 (34/1) | – | 1 (1/0) | 36 (36/0) | |
Phylogenetic and molecular genetic analysis of influenza viruses were performed. Clustering of influenza viruses into clades and subclades was carried out on the basis of HA nucleotide sequences; for comparison, the sequences of vaccine strains, recommended in each season for the Northern Hemisphere, cultured in chicken embryos were used, since such vaccines are widely used in Russia.
Influenza A(H1N1)pdm09 viruses
The distribution of sequenced viruses into genetic clusters (clades, subclades and subgroups) and the results of comparison with the vaccine strain of each season are presented in Table 2. The homology of the nucleotide sequences of the HA gene of the studied viruses and vaccine strains varied in the range of 99.2–97.4% depending on the genetic cluster affiliation. The largest differences (up to 2.6%) were observed in the 2019–2020 season.
Table 2. Results of nucleotide sequence analysis of НА influenza A(H1N1)pdm09 viruses received by the Reference center in 2019–2023
Epidemic season, years | Vaccine strain (genetic cluster) | Number of samples studied | Genetic cluster | HA gene homology with the vaccine strain, % |
2019–2020 | A/Brisbane/02/2018 (6В.1A.1) | 39 | 6В.1A.5a | 98.4–99.2 |
18 | 6В.1A.5a.1 | 98.2–98.6 | ||
1 | 6В.1A.5a.2 | 99.2 | ||
1 | 6В.1A.7 | 98.2 | ||
2022–2023 | A/Victoria/2570/2019 (6В.1A.5a.2) | 351 | 6В.1A.5a.2a | 97.4–98.9 |
Based on sequencing results, A(H1N1)pdm09 viruses from the 2019-2020 season belonged to subclade 6B.1A.5 subgroups 5a, 5a.1, 5a.2, and subclade 6B.1A.7. This season, viruses from subclade 6B.1A.5 subgroup 5a (66%), characterized by amino acid substitutions N129D and T185A in HA1, prevailed. Subgroup 6B.1A.5a.1, characterized by amino acid substitutions D187A, Q189E, included 31% of the studied viruses. One virus belonged to subgroup 6B.1A.5a.2 (amino acid substitutions N156K, L161I, V250A), 1 virus belonged to subclade 6B.1A.7 (amino acid substitutions K302T in HA1 and I77M, N169S, E179D in HA2).
The A/Brisbane/02/2018 strain recommended for inclusion in the vaccine for the 2019–2020 epidemic season for the Northern Hemisphere belonged to subclade 6B.1A.1.
In the 2020–2021 and 2021–2022 seasons, influenza A(H1N1)pdm09 viruses were not sent to the reference center because they were not circulating.
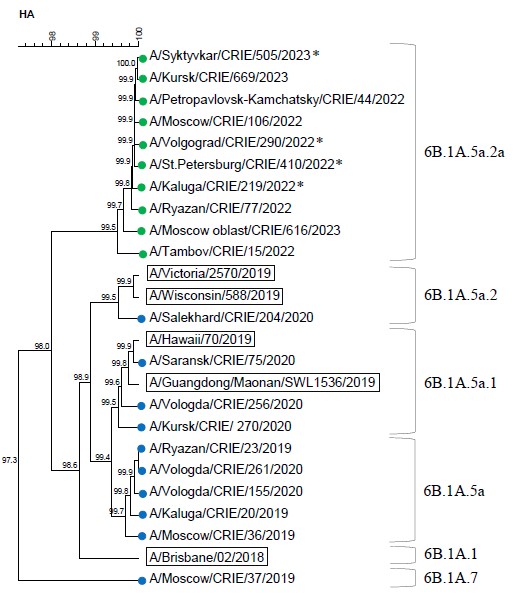
All tested A(H1N1)pdm09 viruses from the 2022–2023 season belonged to clade 6B.1A.5a.2a, characterized by amino acid substitutions K54Q, A186T, Q189E, E224A, R259K, and K308R. This season's vaccine strain A/Victoria/2570/2019 was assigned to subgroup 6B.1A.5a.2.
The results of phylogenetic analysis on the HA gene of influenza A subtype H1N1pdm09 viruses in the 2019–2023 seasons are shown in Figure 2. The dendrogram contains a set of selected sequences of HA, which represented genetic diversity of circulated influenza viruses.

Fig. 2. Dendrogram of the HA gene of influenza A(H1N1)pdm09 viruses (data of the CRIE Reference center, 2019–2020, 2022–2023).
Vaccine strains are indicated by rectangles. Blue and green circles indicate the sequences of viruses circulating in 2019–2020 and 2022–2023 influenza seasons, respectively.
Fatal cases are marked with an asterisk.
Five antigenic sites are identified in the HA molecule of influenza A(H1N1)pdm09 virus, which include the following amino acid positions: Sa (121–122 and 150–162), Sb (184–195), Ca1 (163–167, 200–202 and 232–235), Ca2 (133–139 and 218–219), Cb (67–72) [17].
From 7 to 12 mutations were detected in the HA amino acid sequence of 2019-2020 viruses compared to the vaccine strain A/Brisbane/02/2018. Of these, 7 were located in antigenic sites: Sa — S121I/N (n = 2), N156K (n = 1), L161I (n = 1), Sb — T185I (n = 57), D187A (n = 18), Q189E (n = 16), S190N (n = 1). An R205K substitution was found near the Ca1 antigenic site (n = 2). The mutations D187A (n = 18), R221K (n = 2), and D222N (n = 1) were located in the receptor-binding site. One sample belonged to subclade 6B.1A7 and carried amino acid substitutions E68D (in Cb), T120A, S121N (in Sa1), R223Q, K302T (in HA1), and I77M, N169S, E179D (in HA2).
Influenza A(H1N1)pdm09 2022–2023 viruses relative to the vaccine strain contained 6-12 amino acid substitutions. R259K, K54Q, A186T, Q189E, E224A, K308R/G substitutions in HA1 were detected more frequently. Some of the mutations affected antigenic sites: Sa — S121N (n = 1), S122L (n = 1), K154R (n = 1), G155E (n = 1), N162S (n = 1, loss of glycosylation site), Sb — A186T (n = 351), D187V (n = 1), Q189E (n = 351), N194S (n = 1), Ca1 – V234I (n = 1), Cb — S69P (n = 1), L70F (n = 1). Amino acid substitutions N125H (n = 1), R205K (n = 3) were found adjacent to antigenic sites. The D187V (n = 1) and D222N (n = 12) mutations were located in the receptor-binding site.The D145N mutation in HA2 resulting in an additional glycosylation site was found in 2 virus samples.
In 2019–2020, all influenza A(H1N1)pdm09 viruses lacked mutations conferring resistance to oseltamivir and zanamivir, whereas in 2022–2023 viruses with the H275Y mutation in the NA accounted for 2.3% and were detected in respiratory swabs of unvaccinated patients from the Arkhangelsk region in December 2022. All A(H1N1)pdm09 influenza viruses for which the M gene sequence was obtained were resistant to adamantanes (S31N mutation in M2).
Influenza A(H3N2) viruses
The distribution of the studied influenza A(H3N2) viruses by genetic clusters and the results of comparison with the vaccine strain according to each season are presented in Table 3. The homology of the nucleotide sequences of the HA gene of the studied viruses and vaccine strains varied in the range of 99.0–95.8% depending on the genetic cluster affiliation. Maximum differences (3.6–4.2%) were observed in the 2019–2020 season.
Table 3. Results of НА nucleotide sequence analysis of influenza A(H3N2) viruses received by the Reference center in 2019–2023
Epidemic season. years | Vaccine strain (genetic cluster) | Number of samples studied | Genetic cluster | HA gene homology with the vaccine strain. % |
2019–2020 | A/Kansas/14/2017 (3C.3a.1) | 7 | 3C.2a1b.1b | 96.2–96.4 |
4 | 3C.2a1b.2a | 95.8–96.0 | ||
2020–2021 | A/Hong Kong/2671/2019 (3C.2a1b.1b. previously 3C.2a1b + T135K-B) | 2 | 3C.2a1b.2a.2a.2 | 97.3–97.4 |
2021–2022 | A/Cambodia/e0826360/2020 (3C.2a1b.2a.1a) | 3 | 3C.2a1b.2a.2 | 98.6–98.7 |
3 | 3C.2a1b.2a.2a.1 | 98.6–98.7 | ||
105 | 3C.2a1b.2a.2a.2 | 98.1–98.9 | ||
13 | 3C.2a1b.2a.2c | 98.6–98.9 | ||
2022–2023 | A/Darwin/9/2021 (2a. previously 3C.2a1b.2a.2a) | 2 | 3C.2a1b.2a.2a | 99.4 |
2 | 3C.2a1b.2a.2a.1b | 98.9–99.0 | ||
3 | 3C.2a1b.2a.2a.3a.1 | 98.5–98.6 | ||
3 | 3C.2a1b.2a.2b | 98.5 |
Influenza A(H3N2) viruses from the 2019–2020 season were categorized as group 3C.2a1b, clusters 3C.2a1b.1b and 3C.2a1b.2a. Vaccine strain A/Kansas/14/2017 was assigned to cluster 3C.3a.1 (characterized by amino acid substitutions at positions S91N, N144K, the latter resulting in the loss of a potential glycosylation site, F193S in HA1 and D160N in HA2). Cluster 3C.2a1b.1b, characterized by amino acid substitutions at positions S137F, A138S, and F193S in HA1, included 64% of the samples. The cluster 3C.2a1b.2a, characterized by amino acid substitutions at positions K83E, Y94N in HA1 and I193M in HA2, included 36% of the samples.
In the hemagglutinin molecule of influenza A(H3N2) virus, the following amino acid positions are considered to be antigenic sites:
- A (122, 124, 126, 130–133, 135, 137, 138, 140, 142–146, 150, 152, 168);
- B (128, 129, 155–160, 163–165, 186–190, 192–194, 196–198);
- C (44–48, 50, 51, 53, 54, 273, 275, 276, 278–280, 294, 297, 299, 300, 304, 305, 307–312);
- D (96, 102, 103, 117, 121, 167, 170–177, 179, 182, 201, 203, 207–209, 212–219, 226–230, 238, 240, 242, 244, 246–248);
- E (57, 59, 62, 63, 67, 75, 78, 80–83, 86–88, 91, 92, 94, 109, 260–262, 265) [18].
Three antigenic sites overlap with the receptor-binding site: site A with loop 130 (135, 136, 137, 138, 153); site B with helix 190 (186, 190, 194, 195); and site D with loop 220 (226 and 228) [19].
Both influenza A(H3N2) viruses of the 2020–2021 season belonged to subclade 2a.2 of subgroup 3C.2a1b.2a.2a.2a.2, which is characterized by amino acid substitutions Y159N, T160I (loss of glycosylation site, L164Q, G186D, D190N) in HA1, whereas vaccine strain A/Hong Kong/2671/2019 belonged to cluster 3C.2a1b.1b.
In the 2021–2022 season, as in the previous season, all influenza A(H3N2) viruses belonged to subcluster 3C.2a1b.2a.2. In the 2022–2023 classification, viruses were assigned to 4 subclades: viruses of 3C.2a1b.2a.2a.2 (amino acid substitutions D53G, R201L, S219Y) prevailed, 3C.2a1b.2a.2c (amino acid substitutions S205F, A212T) were less frequently found, and 3C.2a1b.2a.2a.2 and 3C.2a1b.2a.2a.2a.1 (amino acid substitutions D53G, D104G, K276R) were even less frequently found. The vaccine strain A/Cambodia/e0826360/2020 belonged to group 3C.2a1b.2a, but to a different genetic subgroup 3C.2a1b.2a.1a, characterized by amino acid substitutions L157I, K220R.
Influenza A(H3N2) viruses from the 2022-2023 season belonged to clade 3C.2a1b.2a.2a.2, as did the vaccine strain A/Darwin/9/2021, which was in the vaccine administered in the Northern Hemisphere in 2022–2023. Within the clade, the viruses were differentiated into subclades: 3 belonged to 3C.2a1b.2a.2a.3a.1 (amino acid substitution I140K), 3 belonged to 3C.2a1b.2a.2b (characterized by amino acid substitutions E50K, F79V, I140K), and 2 belonged to 3C.2a1b.2a.2a.2a.1b (amino acid substitutions I140K, R299K). Only 2 HAs were close to the vaccine strain A/Darwin/9/2021 (subclade 3C.2a1b.2a.2a, characterized by amino acid substitution H156S).
The results of phylogenetic analysis on the HA gene of influenza A subtype H3N2 viruses are presented in Fig. 3. In the 2019–2020 season, 21–23 mutations were detected in the HA sequence of viruses compared to the vaccine strain A/Kansas/14/2017, 19 of these mutations located in antigenic sites:

Fig. 3. Dendrogram of the HA gene of influenza A(H3N2) viruses (data from the CRIE Reference center, 2019–2023).
Vaccine strains are indicated by rectangles. Blue, violet, red and green circles indicate the sequences of viruses circulating in 2019–2020, 2020–2021, 2021–2022 and 2022–2023 influenza seasons, respectively. Fatal cases are marked with an asterisk.
- A — T131K (n = 3), T135K (n = 7), S137F (n = 7), I140K (n = 7), K144S (n = 10);
- B — A128T (n = 3), S159Y (n = 10), K160T (n = 10), N190D (n = 10), S193F (n = 3);
- D — N121K (n = 10), N171K (n = 10), V230I (n = 1), T246N (n = 10);
- E — E62G (n = 10), K83E (n = 3), N91S (n = 10), K92R (n = 10), Y94N/S (n = 3).
Three substitutions were localized in the receptor-binding site: in loop 130, T135K, S137F, and in helix 190, N190D. The I77V, M149I, and G155E mutations in HA2 of 11 viruses were found.
Mutations that lead to the appearance of new potential N-glycosylation sites were found: A128T (in 3 viruses of subgroup 3C.2a1b.2a), K160T (in all 10 viruses), T246N (in all 10 viruses). Seven viruses of subgroup 3C.2a1b.1b had a T135K mutation that results in loss of the N-glycosylation site.
A(H3N2) viruses examined in the 2020–2021 season, there were 22–24 substitutions in the HA sequences compared to the vaccine strain A/Hong Kong/2671/2019, 16 of which were in the antigenic sites of both viruses: A — T131K, K135T, F137S, S138A; B — A128T, H156S, Y159N, L164Q, V186D, D190N; C — D53G; D — R201K, S219Y; E — K83E, Y94N. One virus had an I214V mutation in site D; 5 substitutions were localized in the receptor-binding site: in loop 130, K135T, F137S, S138A; and in helix 190, V186D, D190N. Additionally, N225D, A128T, and K135T mutations were found outside the antigenic sites in both viruses, the latter resulting in new potential N-glycosylation sites.
In the 2021–2022 season, HA amino acid sequences contained 9–12 mutations compared to the vaccine strain A/Cambodia/e0826360/2020.
The following substitutions in antigenic sites were detected in 124 viruses of all subclades: B, Y159N (1 had Y159S), K160I, L164Q, R186D, D190N, P198S; D, N171K. The R186D, D190N substitutions were in the receptor-binding site. Three viruses of subclade 3C.2a1b.2a.2a.2 had mutations: A — I140K, D — R201I. One virus had the S219Y mutation in the antigenic site D.
Three viruses of subclade 3C.2a1b.2a.2a.1 contained mutations: C — D53G, K276R; B — H156S; outside antigenic sites — D104G; 105 viruses of subclade 3C.2a1b.2a.2a.2a.2 had mutations in antigenic site D — R201K, S219Y. Total 104 viruses had an I25V substitution outside the antigenic sites. Four viruses had mutations that lead to loss of the glycosylation site: 3 viruses had N122D, 1 had N165K. Thirteen viruses of subclade 3C.2a1b.2a.2c had an A212T mutation in antigenic site D. Twelve viruses had an S205F substitution outside the antigenic sites. Two viruses had an S124N mutation resulting in loss of the glycosylation site.
In the 2022–2023 season, the A(H3N2) viruses studied had 2–9 amino acid substitutions in HA compared with vaccine strain A/Darwin/9/2021 and 3–9 substitutions compared with A/Darwin/6/2021 (the sequence of strain A/Darwin/9/2021 differs from A/Darwin/6/2021 by the G53D mutation). Two viruses of subclade 3C.2a1b.2a.2a had a mutation in the D site, I217V. Two viruses of subclade 3C.2a1b.2a.2a.1b had mutations localized in site A — I140K, C — K276R and R299K. Three viruses of subclade 3C.2a1b.2a.2a.3a.1 had mutations in the C site — E50K, D53N, A — I140K, B — I192F; 3 viruses of subclade 3C.2a1b.2a.2b had E50K mutations in the C site and I140K mutations in site A. Two viruses had an amino acid substitution of N96S in HA1 leading to the appearence of N-glycosylation site, and one had an N122D mutation in HA1 leading to loss of the N-glycosylation site.
No resistance mutations to oseltamivir and zanamivir were detected in the NA of influenza A(H3N2) viruses tested for the 2019–2023 seasons, while all had the S31N adamantane resistance mutation in M2.
Influenza B viruses
All influenza B viruses received for study in 2019–2023 belonged to the B/Victoria lineage (according to PCR and sequencing results).
The distribution of investigated influenza B viruses by genetic clusters and the results of comparison with the vaccine strain of each season are presented in Table 4. The homology of the nucleotide sequences of the HA gene of the studied viruses and vaccine strains varied in the range of 99.7–98.2% depending on the genetic cluster affiliation. Maximum similarity (99.0–99.7%) was observed in the season 2022–2023.
Table 4. Results of НА nucleotide sequence analysis of influenza B viruses of the Victoria lineage received by the Reference center in 2019–2023
Epidemic season | Vaccine strain (genetic cluster) | Number of samples studied | Genetic cluster | HA gene homology with the vaccine strain. % |
2019–2020 | B/Colorado/06/2017 (V1A.1. previously 1A(∆2)B) | 55 | V1A.3 | 98.2–98.8 |
1 | V1A.3a.1 | 98.6 | ||
2021–2022 | B/Washington/02/2019 (V1A.3. previously 1A(∆3)B) | 7 | V1A.3a.2 | 98.2–98.7 |
2022–2023 | B/Austria/1359417/2021 (V1A.3a.2) | 104 | V1A.3a.2 | 99.0–99.7 |
In the 2019–2020 season, 98% of influenza B viruses belonged to the Victoria lineage subclade V1A.3 (1A(Δ3)B), characterized by triple deletion of amino acid residues 162–164 and amino acid substitutions K136E, G133R in HA; the remainder belonged to the subgroup V1A.3a.1, characterized by amino acid substitutions in HA1 V117I, V220M. The vaccine strain B/Colorado/06/2017 for the 2019–2020 Northern Hemisphere epidemic season belonged to subclade V1A.1 (1A(D 2)B) of the Victoria lineage, characterized by double deletion of amino acid residues 162–163 and amino acid substitutions at positions D129G, I180V in HA1, R151K in HA2.
In the 2021–2022 season, 7 influenza B viruses belonged to the Victoria lineage subclade V1A.3, subgroup V1A.3a.2 (characterized by amino acid substitutions at HA1 positions A127T, P144L, K203R). Vaccine strain B/Washington/02/2019, which was included in the 2021–2022 Northern Hemisphere vaccines, belonged to the Victoria lineage of subclade V1A.3.
In the 2022–2023 season, all influenza B viruses belonged to lineage Victoria subclade V.1A.3a.2. Vaccine strain B/Austria/1359417/2021, which was included in vaccines in Russia in 2022–2023, also belonged to lineage Victoria subclade V.1A.3a.2. The results of phylogenetic analysis on the HA gene of influenza B viruses of lineage Victoria are presented in Fig. 4.

Fig. 4. Dendrogram of the HA gene of influenza B viruses of the Victoria lineage.
Data from the CRIE Reference center, 2019–2020, 2020–2021, 2022–2023. Vaccine strains are indicated by rectangles. Blue, red and green circles indicate the sequences of viruses circulating in 2019–2020, 2021–2022 and 2022–2023 influenza seasons, respectively. Fatal cases are marked with an asterisk.
There are 4 antigenic sites in the HA molecule of influenza B virus: loop 120 and adjacent regions (116–137), loop 150 (141–150), loop 160 (162–167), helix 190 and its surrounding regions (194–202) [20]. The receptor-binding site is formed by helix 190 (193–202), loop 240 (237–242), loop 140 (136–143) [21].
In the 2019–2020 season, the HA amino acid sequences of viruses had 8–12 mutations compared to the vaccine strain B/Colorado/06/2017, as well as an additional amino acid deletion at position 164. There were 11 substitutions in antigenic sites: I117V (n = 2), R118K (n = 2), N126K (n = 5), A127T (n = 1), E128K (n = 1), D129N (n = 55), G133R (n = 50), Y135D (n = 1), K136E (n = 56) in loop 120; N166D (n = 1) in loop 160; N197D (n = 1) in helix 190. Three substitutions were in the receptor-binding site: in helix 190, N197D; in loop 240, P241Q (n = 1); in loop 140, K136E. One virus each had N166D, N197D, and N233S substitutions that result in loss of the glycosylation site.
Influenza B viruses in the 2021–2022 season had 8–9 amino acid substitutions in HA compared to the vaccine strain B/Washington/02/2019. Amino acid substitutions were located in 5 positions of antigenic sites: in loop 120, A127T (n = 7), R133G (n = 7); in loop 150, P144L (n = 7), N150K (n = 7); and in helix 190, N197D/E (n = 7). One substitution was in the receptor-binding site, in helix 190 – N197D/E. Mutations N197D observed in 6 viruses and N197E identified in 1 virus result in loss of the glycosylation site.
There were 1–6 mutations in the HA of influenza B viruses in the 2022–2023 season compared to the vaccine strain B/Austria/1359417/2021. Seven substitutions were in antigenic sites: in loop 120 — T121N (n = 25), H122N (n = 3), E128K (n = 67), in loop 150 — G149E (n = 1), in helix 190 — D197E (n = 40), T199A/I (n = 100). Two substitutions were in the receptor-binding site, in helix 190 — D197E, T199A/I. In one virus, a T196I substitution was found in HA2 that resulted in loss of the glycosylation site.
No mutations in the NA gene that reduce sensitivity to oseltamivir and zanamivir were found in the examined influenza B viruses of the 2019–2023 seasons.
Discussion
The etiologic structure of influenza varied in different epidemic seasons in Russia and globally. The samples containing influenza viruses submitted to the Reference Center for study were randomly selected in Russian regions during patient screening, so we can assume that their spectrum and genetic characteristics reflect general patterns across the country and allow us to judge the structure of influenza and genetic diversity of circulating viruses in Russia.
In the 2019–2020 season, the Reference Center received mainly influenza A(H1N1)pdm09 and B viruses: 46.8% of A(H1N1)pdm09, 8.7% of influenza A(H3N2) , and 44.5% of B/Victoria. According to the WHO National Influenza Center, all three influenza subtypes were present in the Russian population this season, with influenza B/Victoria virus predominating (43.7%) [22]. According to the European Center for Disease Prevention and Control (ECDC), in the European region, among the viruses that were typed, 51% A(H1N1)pdm09, 40.1% A(H3N2), 8.7% B/Victoria, 0.2% B/Yamagata were detected3. A(H3N2) viruses were the second most abundant, but not B/Victoria influenza viruses as in Russia, which may be due to the different patterns of spread of these virus types in different countries.
In the 2020–2021 season, only 2 influenza viruses (A(H3N2)) were tested in the Reference Center. According to the WHO National Influenza Centre, in Russia, only 37 positive materials were found during the entire season during PCR-based influenza screening, and only 2 influenza B/Victoria viruses were isolated in culture and antigenically characterized [22]. In the European region, this season also saw a 99.4% decrease in the number of confirmed influenza cases compared to the 2019–2020 season [22]. Influenza viruses were distributed as follows: 14.2% A(H1N1)pdm09, 80.6% A(H3N2), 4.9% B/Victoria, 0.3% B/Yamagata [9], i.e., just as in Russia, A(H3N2) virus predominated.
The low activity of influenza viruses worldwide was caused by the emergence of the novel betacoronavirus SARS-CoV-2 in December 2019 in China and subsequent preventive and protective measures [23]. These included restrictions on the movement of people: closing countries' borders, suspending international flights, quarantining those arriving in the country and isolating those who became ill. In addition, personal hygiene practices (frequent hand washing, use of disinfectants and personal protective equipment) were monitored to reduce transmission of influenza viruses. The timing of these measures correlates directly with the sharp drop in influenza incidence in 2020–2021.
In the 2021–2022 season, the number of viruses tested at the Reference Center increased significantly, with influenza A(H3N2) virus predominating (94.7%,) and influenza B viruses accounting for 5.3%. No samples containing influenza A(H1N1)pdm09 virus were received. According to the WHO National Influenza Center, the A(H3N2) subtype was predominantly detected in Russia, with minor involvement of influenza B/Victoria and complete absence of A(H1N1)pdm09 virus [21]. Thus, the etiologic structure of influenza this season in Russia was represented by influenza A(H3N2) virus with a minor contribution of influenza B virus.
According to ECDC data, influenza A(H3N2) virus was also predominant in the European region, with A(H1N1)pdm09 (8.7%) and B being detected much less frequently: A(H3N2) 90.9%, B/Victoria 0.4%, B-Yamagata < 0.1%4. It should be noted that despite reports of isolated findings of B/Yamagata influenza viruses, available databases of genetic information do not contain the HA nucleotide sequences of B/Yamagata influenza viruses detected after March 2020, which means that their affiliation with the Yamagata lineage has not been confirmed by sequencing.
The spectrum of influenza viruses examined in the 2022–2023 season at the Reference Center was as follows: 75.5% influenza A(H1N1)pdm09 viruses, 2.1% influenza A(H3N2) viruses, 22.4% influenza B/Victoria viruses. Among the viruses admitted and examined, 12%, predominantly influenza A(H1N1)pdm09 and to a lesser extent influenza B/Victoria, were viruses found in autopsy material, indicating an increase in influenza severity compared with the 2021–2022 season, when influenza A(H3N2) virus predominated and no autopsy material was admitted (Table 1). The increased number of influenza deaths during seasons of active circulation of influenza A(H1N1)pdm09 virus can be explained by the greater affinity of this influenza A virus subtype for SA-α2,3, which facilitates its spread to the lungs and leads to the development of pneumonia more often. In addition, a lower level of immunity in the population due to lack of exposure to influenza A(H1N1)pdm09 virus during the 2020–2021 and 2021–2022 seasons could have contributed to a more severe epidemic.
According to the WHO National Influenza Center, in Russia as a whole, at the peak of the influenza and acute respiratory viral illness epidemic, influenza viruses were detected in 30% of patient samples examined during PCR screening, with the A(H1N1)pdm09 subtype predominating5. In the 2022–2023 season, according to ECDC data, the structure of influenza in the European region differed from that in Russia: 47.3% — A(H1N1)pdm09, 47.9% — A(H3N2), 4.8% — B/Victoria, 0% — B/Yamagata6, which could be due to the various epidemiological features of the spread of these types of viruses in different countries.
Our data on the distribution of A(H1N1)pdm09 viruses by genetic groups coincided with the data of the A.A. Smorodintsev Research Institute of Influenza: in the 2019–2020 season, the genetic subgroup 6B.1A5 (reference strain A/Norway/3433/2018) prevailed7, in the 2022–2023 season, the genetic subgroup 6B.1A.5a.2 (reference virus A/Sydney/5/2021) prevailed8.
The significance of the detected mutations leading to amino acid substitutions was assessed according to the literature data confirmed experimentally. It should be taken into account that different amino acids differ in physicochemical properties (nonpolar, polar charged and uncharged), they can change the spatial configuration of the protein in different ways, even if the substitution occurred in the same position of the amino acid chain. The functional significance of each mutation must be proven experimentally. In some cases, it is acceptable to draw an analogy, for example, between HAs of subtypes H1 and H5, because HAs of influenza A H1 and H5 viruses belong to the same group (H1/H2/H5/H6/H11/H13), which, according to the predicted amino acid sequence of HAs, may have similar HA spatial conformational structures [25].
N156K and L161I mutations were detected in the antigenic Sa site of influenza A(H1N1)pdm09 virus, both in 1 virus. The S159N mutation in the HA of influenza A(H5N1) virus (position equivalent to N156K in the A(H1N1)pdm09 influenza viruses examined) results in enhanced binding to α-2,6-SA of the ferret respiratory tract mucosa [26]. For the N156K mutation in A(H1N1)pdm09, a change in antigenic properties was predicted in ferret experiments [27].
T185I, D187A, Q189E, and S190N mutations were found in the antigenic site Sb of influenza A(H1N1)pdm09 virus. In HA of influenza A(H1N1) virus, the D187A substitution (190 by H3 numbering) results in decreased binding to α-2,6- and increased affinity for α-2,3-SA [28]. In HA of epidemic influenza A(H1N1) virus, substitution of S190N (193 by H3 numbering) changes the conformation of the Sb antigenic site [29]. An R205K mutation was found near the Ca1 antigenic site of A(H1N1)pdm09 virus. In HA of H5N1 virus, substitution of N224K (numbered H3) at an equivalent position leads to enhanced binding to α-2,6-SA [30].
Near the antigenic Ca2 site of A(H1N1)pdm09 virus, the R221K mutation was found to be part of the HA A(H1N1)pdm09 receptor-binding site. It was shown that changes in this HA can affect the antigenic properties of viruses [31].
The amino acid arginine (R) at position 223 enhances the affinity of A(H1N1)pdm09 for avian-type receptors (α-2,3-SAs). Given the fact that α-2,3-SAs are part of the glycocalyx of the epithelial cells lining human lung alveoli, such mutant strains can cause pneumonia, and further generalized inflammation. By 2020, the majority of circulating influenza A(H1N1)pdm09 viruses (99.80%) had the R223Q mutation, including all the viruses we sequenced, whereas at the early stage of the 2009 pandemic, strains with an arginine (R) in this position were still circulating in a minor population [32]. Apparently, the elimination of mutations that may reduce the spread of viruses due to the high danger of such strains for host life is one of the mechanisms of influenza viruses evolution.
Outside the antigenic sites, 2 viruses of the A(H1N1)pdm09 subtype had the D94N mutation and 1 had the D94E mutation. In HA of influenza A(H5N1) virus, the D94N substitution results in decreased binding to α-2,6-SA and increased affinity for α-2,3-SA, and enhances HA-mediated fusion to the membrane of mammalian cells [33].
The E224A mutation in the receptor-binding site of A(H1N1)pdm09 increases the affinity for α-2,3-SAs (bird-type receptors) localized in human lungs [34].
The D222N mutation in the receptor-binding site also enhances binding to α-2,3-SA. The D222N and D222G mutations are associated with a severe course of influenza, including pneumonia and acute respiratory distress syndrome [35]. Our earlier molecular genetic analysis of influenza A(H1N1)pdm09 viruses circulating in Russia from 2009 to 2014 showed that amino acid substitution of D for G or N at position 222 was statistically significantly more frequently found in the lungs of deceased patients than in respiratory swabs of recovered patients (p < 0.0001 and p = 0.007) [36]. In the 2019–2020 season, the D222N mutation was detected in 1 (2%) virus from bronchoalveolar lavage of a patient with severe community-acquired pneumonia. In the 2022–2023 season, the D222N mutation was present in influenza A(H1N1)pdm09 viruses found in autopsy specimens from 12 patients (22% of viruses from autopsy material; 3% of total).
Our data on the distribution of A(H3N2) viruses by genetic groups matched the data of the Smorodintsev Research Institute of Influenza. In the 2019–2020 season, viruses of genetic subgroups 3C.2 a1b + T131K (reference strain A/South Australia/34/2019) and 3C.2a1b + T135K-B (reference strain A/Hong Kong/2675/2019)9 circulated in the ratio of 2 : 1; in 2021–2022, the genetic subgroup 3C.2a1b.2a.2 prevailed10, and in 2022–2023 — subgroup 3C.2a1b.2a.2 (reference virus Bangladesh/4005/2020)11.
In the 2020–2021 season, no influenza A(H3N2) viruses were isolated at the Smorodintsev Influenza Research Institute12, and the viruses we examined belonged to group 3C.2a1b.2a.2, which was consistent with the ECDC data: influenza A(H3N2) viruses circulating in the influenza A(H3N2) virus population were group 3C.2a1b viruses, most of which were Cambodia- (3C.2a1b.2a.1) and Bangladesh-like (3C.2a1b.2a.2) viruses13.
From 1968 to 2003, antigenic drift of influenza A(H3N2) virus was mainly caused by single mutations in 7 amino acid positions in HA (145 in site A, 155, 156, 158, 159, 189, 193 in site B) near the receptor-binding site [37].
In HA of influenza A(H3N2) virus, substitution A131D (T131K in our samples) increases the charge of the HA molecule and results in decreased neutralization by a monoclonal antibody [38]. The S193R and S193K mutations (S193F in our samples) have been shown to affect the preferential binding of the virus to α2,6 and α2,3 SAs, respectively [39].
In the HA of swine influenza A(H3N2) virus, the S138A substitution (similar to that found in the samples we examined) leads to reduced virus replication in swine respiratory tract epithelial cells that express α2,6 and α2,3 SA receptors [40].
In HA of influenza A(H3N2) virus, substitution K156Q (H156S in our samples) leads to a decrease in the neutralizing antibody activity. This is due to the fact that this amino acid molds the globular head of HA, where it forms a new epitope adjacent to the receptor-binding domain [30].
The data we obtained on the distribution of influenza B viruses by genetic groups also corresponded to the data of the Smorodintsev Research Institute of Influenza: in 2019–2020, the absolute majority of sequenced influenza B viruses belonged to the V1A (del162-164) clade of the Victorian lineage (reference virus B/Washington/02/2019)14; in the 2021–2022 season, influenza B/Victoria viruses of the genetic subgroup V1A.3 a.215 were found, in the 2022–2023 season, influenza type B viruses were assigned to genetic subgroup V1A.3a.2 and similar to the reference virus B/Austria/1359417/2021.
A D197E mutation in the HA receptor-binding site was identified in 40 viruses. The significance of amino acid substitutions in this position of HA was experimentally proved by a group of researchers by passaging strain B/Brisbane/60/2008 in human lung epithelial cell line Calu-3: after 10 consecutive passages, mutation D197T appeared in HA and it was shown that strains with this substitution had significantly lower affinity to SA-α2,3 (bird type). This could be explained by the fact that α2,3-linked glycan forms 2 hydrogen bonds with the amino acid at position 197, and any substitution at this position could affect the binding of HA to receptors [41].
Conclusion
The article presents the results of genetic monitoring of influenza viruses A(H1N1)pdm09, A(H3N2) and B detected in 50 regions of Russia from 2019 to 2023. The results reflect general patterns and allow us to judge the etiologic structure of influenza, the intensity of the epidemic process and the genetic diversity of viruses circulating in Russia.
In the Rospotrebnadzor system, influenza viruses are monitored annually; even in the 2020–2021 season, against the background of the COVID-19 pandemic, screening of patients for influenza by PCR with hybridization-fluorescence detection of amplification products, typing and sequencing of detected influenza viruses continued in the same volume. That is why it can be stated that in Russia against the background of the COVID-19 pandemic in the season 2020–2021 influenza viruses practically disappeared from circulation and appeared again in the season 2021–2022.
The analysis demonstrated the phenomena of continuous evolution with the appearance in each season of genetic variants of influenza viruses A(H1N1)pdm09, A(H3N2) and B that had changes in the HA gene compared to the vaccine strain. Mutations leading to HA amino acid substitutions were recorded in antigenic sites, in the receptor binding region, some of them leading to the formation of new potential glycosylation sites or to their loss.
When comparing influenza A(H1N1)pdm09 viruses circulating in 2022–2023 in Russia with the 2019–2020 vaccine strain A/Brisbane/02/2018, the degree of differences in the nucleotide sequences of the HA gene was 2.7–3.1%, and with the first vaccine strain of influenza A(H1N1)pdm09 virus A/California/07/2009 — 5.0–5.3%.
The degree of difference in the nucleotide sequences of the HA gene of influenza A(H3N2) viruses circulating in 2022–2023 in Russia with the 2019–2020 vaccine strain A/Kansas/14/2017 amounted to 5.3–6.0%.
The level of difference in the nucleotide sequences of the HA gene of influenza B/Victoria viruses circulating in 2022–2023 in Russia with the 2019–2020 vaccine strain B/Colorado/06/2017 was 2.2–2.8%. Influenza B/Yamagata viruses were not identified during the study period.
The highest HA variability was observed for A(H3N2) viruses, which necessitated changing the vaccine strain 3 times in 4 seasons.
Of particular note, all influenza A(H1N1)pdm09 viruses from the 2022–2023 season had the previously unknown E224A mutation in HA, which increases affinity for SA-α2,3 localized in the human lung, which may contribute to complications. The D222N mutation, which is associated with more severe disease, was found in HA in 2% and 3% of influenza A(H1N1)pdm09 viruses in 2019–2020 and 2022–2023, respectively.
Almost all influenza viruses were sensitive to oseltamivir and zanamivir, only 2.3% of influenza A(H1N1)pdm09 viruses in 2022–2023 showed resistance mutation H275Y in NA. In all influenza A(H1N1)pdm09 and A(H3N2) viruses studied, an adamantane resistance mutation S31N in M2 was found.
Our results may help to understand the direction of evolution of influenza viruses. The continuous emergence of mutations in influenza viruses poses a global public health challenge because some mutations provide a selective advantage for viral replication in the upper respiratory tract and human-to-human transmission, and reduce sensitivity to antiviral drugs. Some mutations contribute to a more severe course of influenza and the development of complications. Mutations in antigenic sites allow influenza viruses to evade anamnestic and vaccine-induced antibodies.
Therefore, it is necessary to continue to monitor influenza viruses using molecular genetic analysis, which allows deep differentiation of influenza viruses and determines the trend in the evolution of influenza viruses: the emergence, spread and disappearance from circulation of certain genetic variants.
1 WHO. Influenza (seasonal). URL: https://www.who.int/ru/news-room/fact-sheets/detail/influenza-(seasonal)
2 ECDC. Influenza virus characterisation, Summary Europe, July 2021. Stockholm; 2021. URL: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterisation-summary-europe-july-2021; ECDC. Influenza virus characterization: summary report, Europe, July 2022. Copenhagen; 2022. URL: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterization-summary-europe-july-2022
3 ECDC. Influenza virus characterisation, summary Europe, July 2020. Stockholm; 2020. URL: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterisation-summary-europe-july-2020
4 ECDC. Influenza virus characterisation, Summary Europe, July 2021. Stockholm; 2021. URL: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterisation-summary-europe-july-2021
5 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2023.
URL: https://influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2023&week=39
6 ECDC. Influenza virus characterization: summary report, Europe, February 2023. Copenhagen–Stockholm; 2023. URL: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterization-summary-europe-february-2023
7 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2020.
URL: https://www.influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2020&week=39
8 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2023.
URL: https://influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2023&week=39
9 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2020.
URL: https://www.influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2020&week=39
10 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 24th week of 2022.
URL: https://www.influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2022&week=24
11 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2023.
URL: https://influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2023&week=39
12 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2021.
URL: https://www.influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2021&week=39
13 ECDC. Influenza virus characterisation, Summary Europe, July 2021. Stockholm; 2021. URL: https://www.ecdc.europa.eu/en/publications-data/influenza-virus-characterisation-summary-europe-july-2021
14 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 39th week of 2020.
URL: https://www.influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2020&week=39
15 A.A. Smorodintsev Research Institute of Influenza. Weekly national influenza and ARVI bulletin for the 24th week of 2022.
URL: https://www.influenza.spb.ru/system/epidemic_situation/laboratory_diagnostics/?year=2022&week=24
About the authors
Svetlana B. Yatsyshina
Central Research Institute for Epidemiology
Author for correspondence.
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0000-0003-4737-941X
Cand. Sci. (Biol.), Head, Laboratory of molecular diagnostics and epidemiology of respiratory tract infections
Russian Federation, MoscowAnna A. Artamonova
Central Research Institute for Epidemiology
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0009-0004-6845-9982
laboratory researcher, Laboratory of molecular diagnostics and epidemiology of respiratory tract infections
Russian Federation, MoscowMaria A. Elkina
Central Research Institute for Epidemiology
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0000-0003-4769-6781
researcher, Laboratory of molecular diagnostics and epidemiology of respiratory tract infections
Russian Federation, MoscowAnna V. Valdokhina
Central Research Institute for Epidemiology
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0000-0002-4592-4755
researcher, Scientific group of genetic engineering and biotechnology
Russian Federation, MoscowVictoria P. Bulanenko
Central Research Institute for Epidemiology
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0000-0001-7055-1762
researcher, Scientific group of genetic engineering and biotechnology
Russian Federation, MoscowAleksandra A. Berseneva
Central Research Institute for Epidemiology
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0000-0002-1503-7629
laboratory researcher, Laboratory of molecular diagnostics and epidemiology of respiratory tract infections
Russian Federation, MoscowVasily G. Akimkin
Central Research Institute for Epidemiology
Email: svetlana.yatsyshina@pcr.ms
ORCID iD: 0000-0003-4228-9044
D. Sci. (Med.), Professor, Academician of the Russian Academy of Sciences, Director
Russian Federation, MoscowReferences
- Hause B.M., Collin E.A., Runxia L., et al. Characterization of a novel influenza virus in cattle and swine: proposal for a new genus in the Orthomyxoviridae family. mBio. 2014;5(2):e00031–14. DOI: https://doi.org/10.1128/mBio.00031-14
- Houser K., Subbarao K. Influenza vaccines: challenges and solutions. Cell Host Microbe. 2015;17(3):295–300. DOI: https://doi.org/10.1016/j.chom.2015.02.012
- Osterhaus A.D., Rimmelzwaan G.F., Martina B.E., et al. Influenza B virus in seals. Science. 2000;288(5468):1051–3. DOI: https://doi.org/10.1126/science.288.5468.1051
- Webster R.G., Govorkova E.A. Continuing challenges in influenza. Ann. NY Acad. Sci. 2014;1323(1):115–39. DOI: https://doi.org/10.1111/nyas.12462
- Treanor J. Influenza vaccine — outmaneuvering antigenic shift and drift. N. Engl. J. Med. 2004;350(3):218–20. DOI: https://doi.org/10.1056/NEJMp038238
- Saunders-Hastings P.R., Krewski D. Reviewing the history of pandemic influenza: understanding patterns of emergence and transmission. Pathogens. 2016;5(4):66. DOI: https://doi.org/10.3390/pathogens5040066
- Chang W.K. National influenza experience in Hong Kong, 1968. Bull. World Health Organ. 1969;41(3):349–51.
- Rota P.A., Wallis T.R., Harmon M.W., et al. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology. 1990;175(1):59–68. DOI: https://doi.org/10.1016/0042-6822(90)90186-u
- Vijaykrishna D., Holmes E.C., Joseph U., et al. The contrasting phylodynamics of human influenza B viruses. Elife. 2015;4:e05055. DOI: https://doi.org/10.7554/eLife.05055
- Thyagarajan B., Bloom J.D. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. Elife. 2014;3:e03300. DOI: https://doi.org/10.7554/eLife.03300
- Wei C.J., Boyington J.C., Dai K., et al. Cross-neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci. Transl. Med. 2010;2(24):24ra21. DOI: https://doi.org/10.1126/scitranslmed.3000799
- Job E.R., Deng Y.M., Barfod K.K., et al. Addition of glycosylation to influenza A virus hemagglutinin modulates antibody-mediated recognition of H1N1 2009 pandemic viruses. J. Immunol. 2013;190(5):2169–77. DOI: https://doi.org/10.4049/jimmunol.1202433
- Sriwilaijaroen N., Suzuki Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 2012;88(6):226–49. DOI: https://doi.org/10.2183/pjab.88.226
- Glaser L., Stevens J., Zamarin D., et al. A single amino acid substitution in 1918 influenza virus hemagglutinin changes receptor binding specificity. J. Virol. 2005;79(17):11533–6. DOI: https://doi.org/10.1128/JVI.79.17.11533-11536.2005
- Virk R.K., Jayakumar J., Mendenhall I.H., et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl Acad. Sci. USA. 2020;117(1):619–28. DOI: https://doi.org/10.1073/pnas.1916585116
- Cheng P.K., Leung T.W., Ho E.C., et al. Oseltamivir- and amantadine-resistant influenza viruses A (H1N1). Emerg. Infect. Dis. 2009; 15(6):966–8. DOI: https://doi.org/10.3201/eid1506.081357
- Nobusawa E., Aoyama T., Kato H., et al. Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemagglutinins of influenza A viruses. Virology. 1991;182(2):475–85. DOI: https://doi.org/10.1016/0042-6822(91)90588-3
- Соболев И.А., Курская О.Г., Шаршов К.А. и др. Изменчивость вируса гриппа типа А. Юг России: экология, развитие. 2016;11(1):170–7. Sobolev I.A., Kurskaya O.G., Sharshov K.A., et al. Variability of the influenza A virus. South of Russia: ecology, development. 2016;11(1):170–7. DOI: https://doi.org/10.18470/1992-1098-2016-1-170-177 EDN: https://elibrary.ru/vsasur
- Ndifon W., Wingreen N.S., Levin S.A. Differential neutralization efficiency of hemagglutinin epitopes, antibody interference, and the design of influenza vaccines. Proc. Natl Acad. Sci. USA. 2009;106(21):8701–6. DOI: https://doi.org/10.1073/pnas.0903427106
- Wang Q., Cheng F., Lu M., et al. Crystal structure of unliganded influenza B virus hemagglutinin. J. Virol. 2008;82(6):3011–20. DOI: https://doi.org/10.1128/JVI.02477-07
- Wang Q., Tian X., Chen X., et al. Structural basis for receptor specificity of influenza B virus hemagglutinin. Proc. Natl Acad. Sci. USA. 2007;104(43):16874–9. DOI: https://doi.org/10.1073/pnas.0708363104
- Еропкин М.Ю., Коновалова Н.И., Комиссаров А.Б. и др. Особенности эволюции вирусов гриппа, циркулировавших в России в течение 2-х сезонов, предшествовавших пандемии COVID-19, и в два пандемических сезона. В кн.: Дни вирусологии 2022: Сборник тезисов III Международного форума, посвященного 55-летию со дня основания ФГБУ «НИИ гриппа им. А.А. Смородинцева» Минздрава России. СПб.;2022. Eropkin M.Yu., Konovalova N.I., Komissarov A.B., et al. Features of the evolution of influenza viruses that circulated in Russia during the 2 seasons preceding the COVID-19 pandemic and in two pandemic seasons. In: Days of Virology 2022: Collection of abstracts of the III International Forum dedicated to the 55th anniversary of the founding of the Federal State Budgetary Institution "A.A. Smorodintsev Influenza Research Institute" of the Ministry of Health of Russia. St. Petersburg;2022. EDN: https://elibrary.ru/chdpfr
- Karpova L.S., Stolyarov K.A., Popovtseva N.M., et al. Comparison of the first three waves of the COVID-19 pandemic in Russia in 2020–21. Epidemiol. Vaccine Prev. 2022;21(2):4–16. DOI: https://doi.org/10.31631/2073-3046-2022-21-2-4-16
- Zhu N., Zhang D., Wanget W., et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382(8):727–33. DOI: https://doi.org/10.1056/NEJMoa2001017
- Ha Y., Stevens D.J., Skehel J.J., et al. H5 avian and H9 swine influenza virus haemagglutinin structures: possible origin of influenza subtypes. EMBO J. 2002;21(5):865–75. DOI: https://doi.org/10.1093/emboj/21.5.865
- Wang W., Lu B., Zhou H., et al. Glycosylation at 158N of the hemagglutinin protein and receptor binding specificity synergistically affect the antigenicity and immunogenicity of a live attenuated H5N1 A/Vietnam/1203/2004 vaccine virus in ferrets. J. Virol. 2010;84(13):6570–7. DOI: https://doi.org/10.1128/JVI.00221-10
- Guarnaccia T., Carolan L.A., Maurer-Stroh S., et al. Antigenic drift of the pandemic 2009 A(H1N1) influenza virus in a ferret model. PLoS Pathog. 2013;9(5):e1003354. DOI: https://doi.org/10.1371/journal.ppat.1003354
- Liang Y. Pathogenicity and virulence of influenza. Virulence. 2023;14(1):2223057. DOI: https://doi.org/10.1080/21505594.2023.2223057
- Yoshida R., Igarashi M., Ozaki H., et al. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog. 2009;5(3):e1000350. DOI: https://doi.org/10.1371/journal.ppat.1000350
- Imai M., Watanabe T., Hatta M., et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature. 2012; 486(7403):420–8. DOI: https://doi.org/10.1038/nature10831
- Zhao X.N., Zhang H.J., Li D., et al. Whole-genome sequencing reveals origin and evolution of influenza A(H1N1)pdm09 viruses in Lincang, China, from 2014 to 2018. PLoS One. 2020;15(6):e0234869. DOI: https://doi.org/10.1371/journal.pone.0234869
- Yasugi M., Nakamura S., Daidoji T., et al. Frequency of D222G and Q223R hemagglutinin mutants of pandemic (H1N1) 2009 influenza virus in Japan between 2009 and 2010. PLoS One. 2012;7(2):e30946. DOI: https://doi.org/10.1371/journal.pone.0030946
- Su Y., Yang H.Y., Zhang B.J., et al. Analysis of a point mutation in H5N1 avian influenza virus hemagglutinin in relation to virus entry into live mammalian cells. Arch. Virol. 2008;153(12):2253–61. DOI: https://doi.org/10.1007/s00705-008-0255-y
- Lakdawala S.S., Jayaraman A., Halpin R.A., et al. The soft palate is an important site of adaptation for transmissible influenza viruses. Nature. 2015;526(7571):122–5. DOI: https://doi.org/10.1038/nature15379
- Kilander A., Rykkvin R., Dudman S.G., et al. Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009‐2010. Euro Surveill. 2010;15(9):19498. DOI: https://doi.org/10.2807/ese.15.09.19498-en
- Yatsyshina S., Renteeva A., Deviatkin A., et al. Molecular genetic analysis of the Influenza A(H1N1)pdm09 virus from lethal and recovered cases in Russia from 2009 to 2014: Deletions in the nucleoprotein. Infect. Genet. Evol. 2015;34:160–72. DOI: https://doi.org/10.1016/j.meegid.2015.07.019
- Koel B.F., Burke D.F., Bestebroer T.M., et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013;342(6161):976–9. DOI: https://doi.org/10.1126/science.1244730
- Nakajima S., Nakajima K., Nobusawa E., et al. Comparison of epitope structures of H3HAs through protein modeling of influenza A virus hemagglutinin: mechanism for selection of antigenic variants in the presence of a monoclonal antibody. Microbiol. Immunol. 2007;51(12):1179–87. DOI: https://doi.org/10.1111/j.1348-0421.2007.tb04013.x
- Medeiros R., Naffakh N., Manuguerra J.C., et al. Binding of the hemagglutinin from human or equine influenza H3 viruses to the receptor is altered by substitutions at residue 193. Arch. Virol. 2004;149(8):1663–71. DOI: https://doi.org/10.1007/s00705-003-0287-2
- Busch M.G., Bateman A.C., Landolt G.A., et al. Identification of amino acids in the HA of H3 influenza viruses that determine infectivity levels in primary swine respiratory epithelial cells. Virus Res. 2008;133(2):269–79. DOI: https://doi.org/10.1016/j.virusres.2008.01.014
- Ilyushina N.A., Lee N., Lugovtsev V.Y., et al. Adaptation of influenza B virus by serial passage in human airway epithelial cells. Virology. 2020;549:68–76. DOI: https://doi.org/10.1016/j.virol.2020.08.004
Supplementary files

