


Application of the pseudovirus-based neutralization assay in the search for new antiviral drugs
- Authors: Karpenko L.I.1, Rudometova N.B.1, Nizolenko L.F.1, Loktev V.B.1
-
Affiliations:
- State Research Center of Virology and Biotechnology "Vector"
- Issue: Vol 102, No 4 (2025)
- Pages: 482-494
- Section: REVIEWS
- URL: https://microbiol.crie.ru/jour/article/view/18814
- DOI: https://doi.org/10.36233/0372-9311-684
- EDN: https://elibrary.ru/VRKCFO
- ID: 18814

Cite item

Abstract
In recent years, significant progress has been made in the field of drug development, particularly due to the use of computer modeling methods. One of the key stages in the development of new antiviral drugs is testing the efficacy of promising candidates in in vitro experiments using target viruses. The application of new technologies for conducting primary screening with pseudotyped viruses simplifies research, increases its efficiency and ensures the biosafety of the conducted studies.
The aim of this review is to analyze previous studies that have demonstrated the successful use of pseudovirus technology for the search of new chemotherapeutic agents against a range of RNA-containing viruses.
The analysis involved the literature presented in the PubMed, Scopus, Elsevier, and Google Scholar databases as of March 1, 2025. For the search, the following keywords were used: pseudovirus, virus inhibition, antiviral drugs, RNA viruses.
Pseudotyped viruses are recombinant viral particles that have the core proteins of one virus and the surface proteins of another, studied virus. The advantages of pseudovirus technology are its safety, high level of reproducibility of results, and the possibility of standardization. The lentivirus-based system was one of the first to be developed and remains one of the most in-demand. Using pseudoviruses, candidate molecules for infections caused by RNA-containing viruses, such as HIV-1, hepatitis C virus, tick-borne encephalitis virus, avian influenza viruses, and SARS-CoV-2, have been selected and studied. Most of the selected drugs act at the initial stage of the virus entry into the target cell. The examples provided illustrate the significant contribution of pseudovirus technology in dealing with serious socially significant diseases caused by RNA-containing viruses.
Keywords
Full Text
Introduction
The search and development of new drugs for the prevention and treatment of viral infections is one of the most important objectives in medical chemistry, biology and medicine due to the widespread prevalence of various socially dangerous viral infections and the constant emergence of new viral diseases [1, 2].
Progress in the development of pharmaceuticals in recent years has largely been made possible by the application of computer modeling methods to predict the structure of target molecules and their interactions with candidate drug compounds [3]. The awarding of the Nobel Prize in 2024 for the development of methods for computer-aided protein design and prediction of protein quaternary structure vividly illustrates the significance of scientific achievements in this field of knowledge. In a short period, more than 200 million protein structures were deciphered (predicted) using the AlphaFold2 program, involving approximately 2 million researchers from 190 countries1. Recent advancements in artificial intelligence also create fundamentally new opportunities for designing new drugs based on computer-aided design of candidate drug compounds and target proteins [3].
At the same time, the promising candidates for future antiviral drugs selected using computer technologies need to be tested in real in vitro experiments, followed by in vivo testing of the selected active molecules. Among socially significant infectious diseases, it is important to note infections caused by various viral agents such as HIV-1, hepatitis C, various pathogenic orthoflaviviruses, influenza viruses, SARS-CoV-2, as well as many other dangerous and particularly dangerous infections. Experimental work with these pathogens requires special working conditions to meet strict biosafety requirements and is characterized by the complexity of conducting laboratory experiments, the lack of simple and safe laboratory methods for working with infectious agents, and the inability to cultivate multiple viruses in the laboratory, which makes conducting experiments complicated or even impossible. A fundamentally important alternative for primary screening is the use of pseudotyped viruses [4]. The use of replication-deficient pseudoviruses carrying viral envelope proteins is a safe and useful method widely employed by virologists for studying, screening and developing new antiviral chemotherapeutics.
The aim of the review is to analyze studies that have demonstrated the successful use of pseudovirus technology for the search of new chemotherapeutic agents against a range of RNA-containing viruses.
The analysis included literature presented in the scientific databases PubMed, Scopus, Elsevier, and Google Scholar as of March 1, 2025. For the search, the following keywords were used: pseudovirus, virus inhibition, antiviral drugs, RNA viruses, псевдовирус, ингибирование вируса, противовирусные препараты, РНК-содержащие вирусы.
In the PubMed scientific electronic database, a search using a combination of keywords found 293 sources, of which 228 had full text available in open access. Similarly, the search was also conducted using the Scopus, Elsevier and Google Scholar scientific databases, but for these, the publication date of the articles was limited to 2023–2024 in order not to overlook the most relevant research. Overall, during the literature search in the listed databases in Russian and English, conducted with consideration of selection criteria such as publication year and availability of publications for reading, approximately 1700 sources relevant to the topic were analyzed. Due to the volume limitation for the study, 68 of those sources were selected.
Pseudoviruses
Pseudoviruses are artificially created viruses with defective genomes that, due to similar conformational structures of surface glycoproteins, are capable of entering susceptible cells just like natural viruses do [5–7]. Unlike viruses, pseudoviruses can usually replicate only during a single replication cycle [7, 8]. The replication limitation makes conducting experiments with them safe and provides new opportunities for studying highly pathogenic RNA viruses. It is important to note that the use of pseudoviruses allows for research even in cases where natural viruses are impossible or very difficult to cultivate in laboratory conditions [9].
Once again, it is important to emphasize that, unlike infectious viruses, working with pseudoviruses is safe because changes (mutations) have been incorporated into the coding regions of the genome, limiting the virus's development to just one replication cycle. Therefore, pseudoviruses are often referred to as single-cycle infection viruses. As a rule, pseudotyped viruses carry marker genes (such as luciferase or GFP), which allows for easier and more accurate quantitative assessment in experiments with them.
Thus, the advantages of pseudovirus technology are safety, a high level of reproducibility of results, the possibilities of standardization and the generation of new pseudovirus variants for further research development. This predetermines the widespread use of this technology by researchers to study the characteristics of virus entry into target cells, determine the presence of virus-neutralizing antibodies, as well as search for and develop new antiviral drugs.
Features of pseudovirus construction
Most commonly, pseudoviruses are divided into three main types: those with a lentiviral genome (HIV-1), those with a vesicular stomatitis virus genome and those with a mouse leukemia virus genome [10]. The system based on lentiviruses, particularly HIV-1 Env-pseudoviruses, was one of the first developed for analyzing the immune response to HIV-1 vaccines and searching for antiviral drugs [11]. It remains one of the most sought-after and frequently used in research.
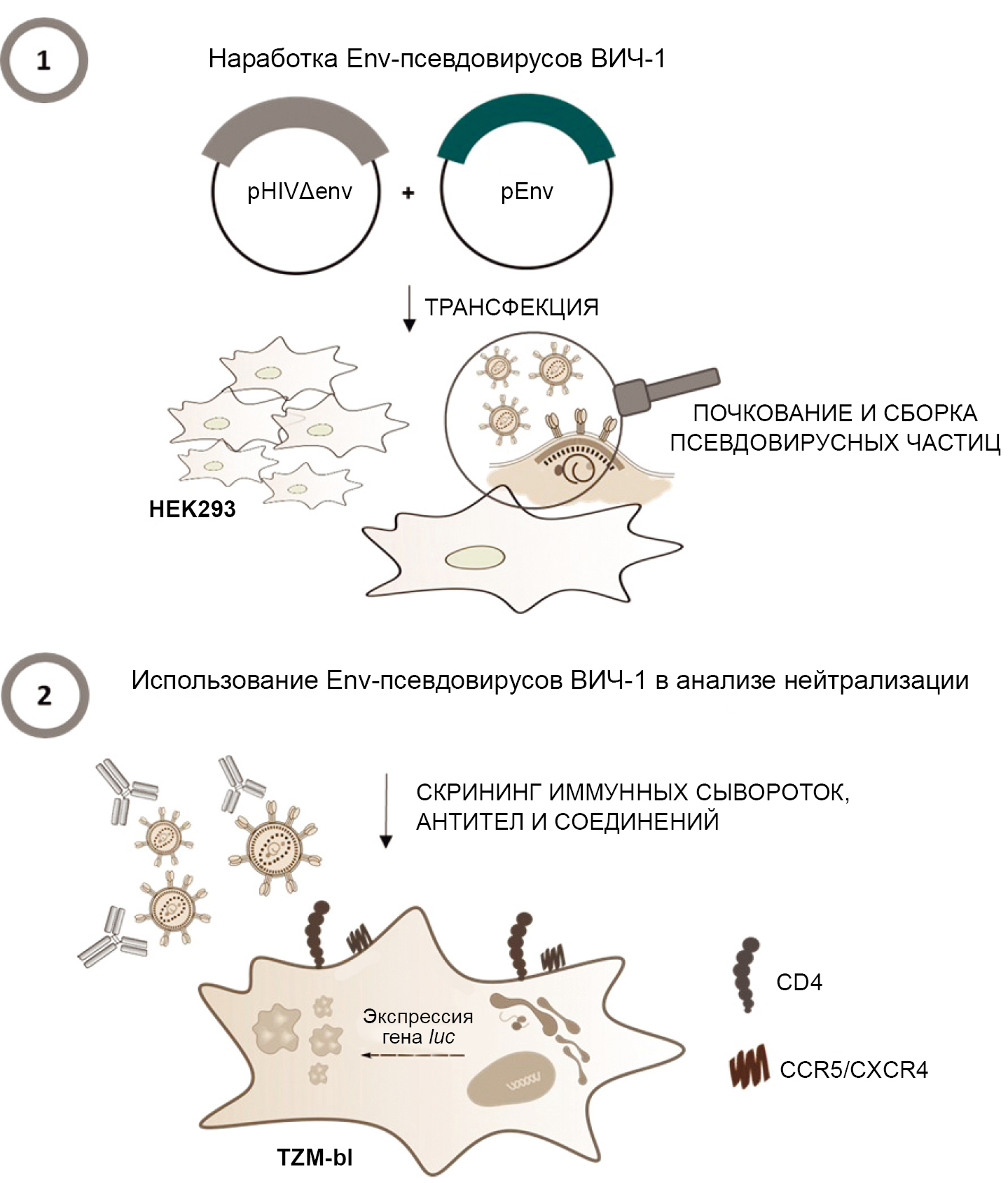
Env-pseudoviruses of HIV-1 are recombinant viral particles obtained by transfecting eukaryotic cells with two plasmids: a core plasmid and an envelope plasmid. The core plasmid contains the genes of structural, regulatory, and accessory proteins of HIV-1 necessary for the assembly of viral particles, as well as sequences required for packaging the viral RNA (Ψ); the envelope plasmid, in turn, carries the gene of the surface glycoprotein (env) of a specific subtype of HIV-1. As a result of transfection, viral particles with a defective genome are formed, which are unable to ensure the assembly of infectious progeny virions upon infection [12, 13]. When working with HIV-1 Env-pseudoviruses, the genetically modified TZM-bl cell line, a derivative of the HeLa cell line, is used. This cell line expresses CD4 receptors and the CCR5 and CXCR4 co-receptors on its surface, which HIV-1 uses to enter target cells. An important feature of the TZM-bl cell line is the presence of integrated reporter genes for firefly luciferase and Escherichia coli β-galactosidase under the transcriptional control of the long terminal repeat of HIV-1, the protein products of which are used to detect the entry of HIV-1 Env-pseudoviruses into target cells. Thus, upon the entry of the pseudovirus into the TZM-bl target cell, the expression of reporter genes is initiated in response to the synthesis of the viral protein Tat, which, for example, for the luciferase gene, is detected using a luminometer. In this case, a high intensity of luminescence corresponds to the entry of pseudovirus particles into target cells, while a suppression of luminescence, on the contrary, indicates the neutralization of HIV-1 Env pseudoviruses [14, 15]. The Figure illustrates the general principle of operation of HIV-1 Env-pseudoviruses.

The general principle of operation of HIV-1 Env-pseudoviruses [29, with added changes].
Practical work with HIV-1 Env-pseudoviruses includes two main stages: Stage 1 — assembly of viral particles using transfection of the HEK293 cell line with two plasmids: core (pHIVΔenv) and envelope (pEnv); Stage 2 — neutralization assay using chemotherapeutic agents to determine their ability to block the entry of pseudoviruses into the target cell.
By replacing the env gene in the lentiviral system with the envelope protein coding genes of other viruses, it is possible to obtain lentiviral particles that display the corresponding viral proteins on their surface. Using this technology, pseudoviruses of enveloped RNA viruses such as influenza viruses [16], coronaviruses [3], retroviruses [17], flaviviruses [18] and others have been obtained, which are successfully used in research.
The pseudovirus platform can equally be useful for studying the entry of viruses that rapidly accumulate mutations due to adaptation in culture, due to the absence of selective pressure such as host immune responses that exist in vivo. For example, in vitro studies using live HIV-1 led to mutations in the Env glycoprotein due to the error-prone nature of reverse transcriptase, which facilitated adaptation to tissue culture and resulted in phenotypic changes. As a result, the entry of tissue culture-adapted HIV-1 strains into cells did not correspond to what occurs during natural infection in humans and, thus, did not provide a clinically relevant model [20].
It should be noted that, despite the aforementioned advantages, the pseudovirus system has a number of limitations that should be considered when conducting any research. This system is primarily designed to simulate the process of viral agent entry into a cell in vitro; however, the process of proliferation and release of viral particles cannot be modeled. The distribution, conformation, and density of heterologous viral glycoproteins on pseudoviruses do not always reflect their natural state on the surface of native viral particles [13].
Therefore, the results of analyses using pseudotyped viruses do not always align with data obtained through analyses using native viruses [13]. Finally, the use of pseudoviruses is effective in studying enveloped viruses. For viruses such as rotavirus and poliovirus, the pseudovirus system functions unsatisfactorily [7]. To address the latter issue, a self-organizing pseudovirus system was developed, but it will not be considered here.
This review will analyze and summarize the results of using pseudovirus technology to search for drugs against HIV-1, orthoflaviviruses and avian influenza viruses.
Pseudoviruses and antiviral drugs
Antiviral drugs that inhibit HIV entry (Riboviria, Pararnavirae, Artverviricota, Revtraviricetes, Ortervirales, Retroviridae, Orthoretrovirinae, Lentivirus)
Currently, the basis of therapy for patients with HIV infection is antiretroviral therapy (ART) — a combination of several antiviral drugs that suppress the replication of the virus by targeting different stages of its life cycle, thereby preserving the immune functions of the body and reducing the risk of virus transmission [21]. However, the spread of HIV resistance to ART reduces the effectiveness of treatment and increases mortality from HIV/AIDS [22].
Targeting the initial stage of the virus's entry into the target cell offers several advantages compared to other stages of the HIV-1 life cycle [23]. Firstly, the viral genetic material cannot integrate into the host cell's genome. Secondly, entry inhibitors do not need to cross the cell membrane, unlike reverse transcriptase, integrase, or protease inhibitors. Thirdly, since the virus entry consists of separate stages, there are several targets for entry inhibitors, which guarantees protection against cross-resistance [24].
An important point in the search for such drugs is that HIV-1 requires co-receptors to enter the cell, which expands the list of targets for antiviral drugs [25]. Among the first effective inhibitors of HIV-1 entry was the drug maraviroc [26]. The drug binds to CCR5, thereby blocking the subsequent stages of viral and cellular membrane fusion and, consequently, the virus's entry into target cells. In this case, HIV-1 cannot enter human macrophages and T-lymphocytes. However, maraviroc can cause serious, life-threatening side effects. This fact once again underscores the necessity for the development of new HIV-1 entry inhibitors.
The pseudovirus technology is widely used to study drugs that block the binding of the virus to membrane receptors and its entry into target cells. The list of such drugs is provided in Table 1.
Enfuvirtide (T20), which for a long time remained the only viral fusion inhibitor used in combination therapy for HIV infection, is a peptide drug, but it has relatively low antiviral activity and easily induces drug resistance. The activity of lipopetide inhibitors developed based on T20 is significantly higher. Their development, structural analysis, function, and comparison of activity in suppressing the entry of HIV-1 and pseudovirus into cells are the focus of the research [27].
Table 1. Antiviral drugs against HIV-1, obtained and/or studied using pseudovirus technology
Substance | Mechanism of action | Reference |
Lipopeptides based on T20 (enfuvirtide) | Fusion inhibitor | [27] |
HNG-105 — modification of the linear peptide 12p1 (RINNIPWSEAMM) | Inhibition of gp120 by interacting with a site distinct from the CD4 or coreceptor binding site, resulting in a sharp decrease in gp120's affinity for either | [28] |
RNA aptamers | Binding to glycoproteins gp120 or gp41 and other viral surface molecules | [29] |
DNA triplexes with hydrophobic modifications | Interaction with the primary pocket in the repeat of N-heptad of glycoprotein 41 (gp41) | [30] |
Thiolated derivatives of pyrimidine | Selective inhibition dependent on the co-receptor preference of the pseudovirus | [31] |
Small molecules NBD-14009 and NBD-14010 are analogs of N-phenyl-N'-(2,2,6,6-tetramethylpiperidin-4-yl)oxalamide | Inhibition of cell fusion and HIV-1 transmission from cell to cell | [32] |
Methylgallate from the edible mushroom Pholiota adiposa | Inhibition of HIV-1 replication in TZM-BL cells infected with a pseudovirus, blocking both the virus entry process and the activity of key enzymes necessary for its life cycle | [36] |
DAVEI is a recombinant peptide chimera of cyanovirin-N (CVN) lectin and HIV-1 gp41 | Binding of gp120 and gp41 | [40] |
Catechin from Peltophorum africanum | Not described | [37] |
Methanol extract of Elaeodendron transvaalense root | Not described | [37] |
Percolation extract of Spatholobus suberectus Dunn | Interaction with the viral envelope glycoprotein gp160 | [38] |
Glycyrrhizin – mixture of derivatives of nicotinic and glycyrrhizic acids | Prevents the virus from entering the target cell | [34] |
Another example could be the artificial peptide HNG-105, created by click conjugation of the linear peptide 12p1 (RINNIPWSEAMM). HNG-105 was studied using surface plasmon resonance spectroscopy and pseudovirus inhibition assays. The results show that the HNG-105 molecule may be effective against HIV-1 subtypes and highlight its potential as a lead for the development of therapeutic and microbicidal agents to help combat the spread of AIDS [28].
HIV entry inhibitors have also been developed based on nucleic acids. Thus, 23 clones of RNA aptamers against the pseudotyped virus HIV-1CAP45, which belongs to subtype C viruses endemic to countries in sub-Saharan Africa and responsible for the majority of HIV-1 infections worldwide, were isolated and studied. Aptamers inhibited the infection of target cells by binding to both different glycoprotein molecules (gp120 or gp41) and other viral surface molecules necessary for infection. Thus, they can be used as analytical tools to study the mechanisms of HIV-1 entry and as inhibitors of this process [29]. Another category of HIV-1 inhibitors may consist of complexes based on DNA triplexes with hydrophobic modifications, interacting with the primary pocket in the N-heptad repeat of glycoprotein 41 (gp41). Using Env-pseudoviruses, it has been demonstrated that triplexes are inhibitors of virus-cell fusion [30].
As potential inhibitors of HIV-1 entry, synthetic chemical compounds were also investigated. Thiolated pyrimidine derivatives were synthesized and their antiretroviral effect against human immunodeficiency virus type 1 (HIV-1IIIB) and chimeric HIV-1 pseudovirions was quantitatively determined in viral infectivity assays, including syncytium inhibition assays, as well as single-cycle viral infection assays on HeLaCD4-LTR/β-gal cells. The inhibition was selective and depended on the co-receptor preference of the pseudovirus [31].
Through targeted screening of commercial libraries in 2005, small molecules, analogs of N-phenyl-N'-(2,2,6,6-tetramethylpiperidin-4-yl)-oxalamide, were identified as a new class of HIV-1 entry inhibitors that blocked the gp120-CD4 interaction [32]. However, they had a number of significant drawbacks. The following decade, this group of scientists systematically studied and modified these substances, ultimately resulting in the small molecules NBD-14009 and NBD-14010, which were tested against a panel of 51 HIV-1 Env-pseudoviruses representing various subtypes of clinical isolates. These compounds exhibited antiviral activity in the nanomolar range (IC50 ≈ 150 nM). They also inhibited cell fusion and the transmission of HIV-1 from cell to cell [33].
Studies dedicated to natural compounds targeting the stage of HIV-1 entry into target cells are of particular interest. The drug Glycyvir — a multi-component mixture containing mono-, di-, tri- and tetranicotinates of glycyrrhizic acid — exhibited pronounced inhibitory activity against HIV-1 pseudoviruses of subtypes B, A6, and the recombinant form CRF63_02A (IC50 range 3.9–27.5 µM). Analysis of the inhibitory activity of Glycyvir depending on the timing of its addition to HIV-1 Env-pseudoviruses and TZM-bl cells suggested that this drug acts at the stage of virus entry into the target cell [34]. Later, a modified synthesis method for the glycyrrhizin preparation was developed, which allowed for the replacement of the initial reagents with less toxic and cheaper ones, resulting in a glycyrrhizin preparation with biological activity similar to the original Glycyvir [35].
In the study [36], the antiviral activity of methyl gallate from the edible mushroom Pholiota adiposa was demonstrated. Methyl gallate inhibited the entry of Env-pseudovirus into TZM-bl cells, while exhibiting low toxicity towards the cell cultures used [36]. To identify inhibitory substances from plants, an HIV-1 subtype C pseudovirus (HIV-1-C) was created, and as controls, wild-type HIV-1 subtype B pseudoviruses (HIV-1-B) and mutants resistant to nucleoside and non-nucleoside reverse transcriptase inhibitors were used. Thus, catechin obtained from Peltophorum africanum inhibited the entry of HIV-1-C and HIV-1-B pseudoviruses with selectivity indices of 6304 µM (IC50: 0.49 µM, CC50: 3089 µM) and 1343 µM (IC50: 2.3 µM, CC50: 3089 µM), respectively [37]. The percolation extract of Spatholobus suberectus Dunn (SSP) possesses a broad spectrum of antiviral activity against the entry of SARS-CoV, H5N1 IAV, and HIV-1. In particular, in the case of HIV-1, SSP interacted with the viral envelope glycoprotein gp160, which is responsible for the virus's entry into the host target cells [38].
New antiviral drugs capable of inactivating the infectious activity of viral particles have been named inactivators. Unlike fusion inhibitors and receptor antagonists, they can actively inactivate virions in the blood by interacting with one or more sites on the viral envelope glycoproteins. It is suggested that a number of developing protein- and peptide-based virus inactivators may be safe for use in human treatment [39]. For example, a recombinant chimera designated as DAVEI (dual-acting virucidal entry inhibitor), consisting of cyanovirin-N (CVN) lectin and a peptide from the conserved membrane-proximal external region of HIV-1 gp41 envelope protein (MPER), could effectively inactivate HIV-1 Bal.01 pseudovirus with an EC50 value of 28.3 nM. Irreversible inactivation of HIV-1 virions occurred through dual interaction with gp120 and gp41. At the same time, the original CVN or MPER molecules themselves are not capable of inactivating HIV-1 virions [40].
Antiviral drugs that inhibit orthoflaviviruses
Another family of RNA-containing viruses with immense medical significance is the viruses belonging to the family Flaviviridae (Riboviria, Orthornavirae, Kitrinoviricota, Flasuviricetes, Amarillovirale, Flaviviridae). Today, the family Flaviviridae includes more than 90 species of viruses and many new, yet unclassified viruses. It is important to note that currently, orthoflaviviruses are almost globally distributed, and hundreds of millions of people encounter them annually, many become ill, and some even die. It is customary to distinguish the so-called major flavivirus infections, which are caused by the dengue, Japanese encephalitis, West Nile, yellow fever and Zika viruses.
The hepatitis C virus (HCV), which also belongs to a separate genus within the family Flaviviridae, is responsible for approximately 170 million human infections. The disease often progresses in a chronic form, ending tragically, and the hepatitis C virus has quite rightly earned the unofficial title of silent killer [41]. This is the only representative of orthoflaviviruses for which officially registered highly effective drugs exist, leading to the cure of patients in most cases. These include the HCV protease inhibitors boceprevir and telaprevir [42], as well as daclatasvir, sofosbuvir and simeprevir, which target viral enzymes — NS5 polymerase or NS3 protease. They provide highly effective antiviral therapy for the overwhelming majority of patients [43]. However, in certain cases, such therapy is accompanied by adverse side effects [44]. Therefore, the search for drugs against the hepatitis C virus remains a relevant issue. Thus, a number of studies have been dedicated to substances targeting the hepatitis C virus. It has been demonstrated that harzianic acids A and B, isolated from the Trichoderma harzianum fungus, can affect the HCV envelope protein E1/E2 as well as the host cell CD81, inhibiting the entry of pseudoviruses into cells and exhibiting low cytotoxicity [45]. In the same HCVpp model, it was shown that the plant alkaloid berberine exhibits antiviral activity by interacting with the HCV envelope glycoproteins E1 and E2 [46]. Using pseudotyped HCV virus, it was found that LUMS1 — a modified form of the microvirin lectin, known as an HIV-1 entry inhibitor — can equally effectively inhibit the entry of HCVpp into host target cells [47]. Pangenotypic entry of HCV pseudoparticles into human Huh7 hepatoma cells was inhibited by derivatives of schisandrin acid, a triterpenoid from the Schisandra sphenanther fruits, preventing the fusion of the virion and the cell membrane without exhibiting significant cytotoxicity. These compounds also demonstrated strong antitumor activity against the Bel7404 and SMMC7721 cell lines [48]. Several flavonoid derivatives, in which triazole groups were combined with a pyranoflavonoid scaffold, inhibited the infection of Huh7 cells by the hepatitis C virus. Additional studies on the mechanism of action using pseudoviruses confirmed that the most effective of these compounds inhibited the virus's entry into the cell [49]. It is important to note that the hepatitis C virus is not actually cultivated in laboratory conditions on cell cultures. The successes in the development of antiviral drugs against HCV have largely been predetermined by the development and use of pseudovirus technologies for assessing the antiviral activity of candidate compounds.
Pseudotyped particles have also been created for other viruses of this family, predominantly using a lentiviral system, namely: infectious pseudotyped hepatitis C virus HCVpp carrying unmodified HCV E1 and E2 glycoproteins [50], pseudotyped Japanese encephalitis viruses [51, 52], pseudotyped dengue viruses D2(HIVluc) [53] and Zika — ZikaEnv/HIV-1 [54].
Pseudotyped viruses have been widely used to study receptor interactions between the surface proteins of orthoflaviviruses and host cells, as well as to search for and investigate the mechanism of action of antiviral drugs [55]. The list of such drugs is provided in Table 2.
T. Pan et al. showed that several non-steroidal anti-inflammatory drugs (NSAID), including aspirin, ibuprofen, naproxen, acetaminophen, and lornoxicam, effectively suppress the entry of pseudotyped Zika Env/HIV-1 viruses, as well as Zika virus replication in cell lines and in primary human fetal endothelial cells [54]. Interestingly, the NSAID-inhibitory effect was mediated by the effective reduction of the expression of the AXL cellular protein, a cofactor for Zika virus entry. Thus, a new mechanism of action for antiviral compounds has been described, which involves blocking the entry of the Zika virus by degrading the cofactor that facilitates the virus's entry into the cell. The authors concluded that NSAID could be used in practice to prevent Zika virus infection in pregnant women, as some NSAID, including ibuprofen and acetaminophen, are considered clinically safe.
Table 2. Antiviral drugs against flaviviruses, obtained and/or studied using pseudovirus technology
Substance | Infection | Mechanism of action | Reference |
NSAID (aspirin, ibuprofen, naproxen, acetaminophen, lornoxicam) | Zika | Reduction of cellular AXL protein expression, a cofactor for ZIKV entry | [54] |
Garcianic acids A and B from Trichoderma harzianum | Hepatitis C* | Impact on the E1/E2 viral envelope protein, as well as CD81 of host cells | [45] |
Berberine (plant alkaloid) | Hepatitis C | Interaction with glycoproteins E1 and E2 | [46] |
LUMS1 — a modified form of microvirin (lectin) | Hepatitis C | Interaction with glycoproteins E1 and E2 | [47] |
Derivatives of schisandronic acid (triterpenoid from the Schisandra sphenanthera fruits) | Hepatitis C | Prevent the fusion of the virion and the cell membrane | [48] |
Note. * The pseudovirus is assembled based on the genome of the vesicular stomatitis virus. All other pseudoviruses are constructed based on the HIV-1 genome.
Antiviral drugs that inhibit influenza virus entry
Influenza viruses are highly contagious respiratory pathogens of humans, belonging to the family Orthomyxoviridae (Riboviria, Orthornavirae, Negarnaviricota, Polyploviricotina, Insthoviricetes, Articulavirales, Orthomyxoviridae). The family contains four genera of segmented RNA viruses: Alphainfluenzavirus, Betainfluenzavirus, Deltainfluenzavirus, and Gammainfluenzavirus. In fact, each genus includes one virus species: influenza A virus (Alphainfluenzavirus influenza), influenza B virus (Betainfluenzavirus influenza), influenza C virus (Gammainfluenzavirus influenzae), and influenza D virus (Deltainfluenzavirus influenza). Human diseases are mainly associated with influenza viruses A, B and C, although the family Orthomyxoviridae includes 5 more genera of viruses.
On the lipid envelope of influenza viruses, two main membrane glycoproteins dominate: hemagglutinin (HA) and neuraminidase (NA). The surface glycoprotein HA is responsible for the attachment of the virus and its entry into host cells through sialic acid receptors, while NA, through its enzymatic activity, ensures the release of viral progeny from the infected cell. Subtypes (serotypes) of the influenza A virus are commonly classified in combinations of 18 HA types and 11 NA types [56].
In addition to vaccination, antiviral drugs are used for the treatment and prevention of influenza A virus (IAV) infections. The search for such drugs and the study of their mechanisms of action using pseudoviral particles are the subjects of a number of studies. The list of such drugs is provided in Table 3.
Table 3. Antiviral drugs against influenza A viruses, developed and/or studied using pseudovirus technology
Substance | Influenza strain | Mechanism of action | Reference |
Small molecule TBHQ | H7 A/Netherlands/219/2003, H3 A/Brisbane/10/2007 | The aromatic ring of TBHQ has extensive contact with the HA stem loop region | [59] |
C12-KKWK and C12-OOWO are membrane-active lipopetides | A/Puerto Rico/8/34, A/Aichi/2/68 | Interaction with the HA2 subunit | [60] |
Quercetin (vitamin P) | A/Anhui/1/2005A, A/Xinjiang/1/2006, A/Hong Kong/156/1997, A/Qinghai/59/2005, A/Thailand/Kan353/2004, A/VietNam/1194/2004 | Interaction with the HA2 subunit of the influenza A (H5N1) virus, which mediates the fusion of the viral envelope with the endosomal membrane at the early stage of infection | [61] |
Oligothiophene compounds | H5N1 | Binding to HA | [62] |
H5N1-A/Thailand/ Kan353/2004 | Binding to HA | [63] | |
Pentacyclic triterpene saponins C-28 | A/Duck/Guangdong/99 | Binding to HA | [64] |
Percolation extract of Spatholobus suberectus Dunn | H5N1Turkey | Прямое связывание гликопротеинов вирусной оболочки Direct binding of viral envelope glycoproteins | [38] |
Griffithsin from red algae, and its modification GL25E | H1N1: Puerto Rico/8/1934, California/07/2009, Shanghai/37T/2009, WSN/1933. H3N2: Guizhou/54/1989 | Binding to HA at the stage of virus entry | [65] |
Lentiviral pseudovirus systems have been developed for SARS-CoV, SARS-CoV-2, and avian influenza H5 [57]. However, two other platforms — the vesicular stomatitis virus genome and the mouse leukemia virus genome — are also used in research [58].
A. Antanasijevic et al., analyzing the entry of a pseudovirus based on HIV-1, found that the small molecule tert-butylhydroquinone (TBHQ) inhibits the entry of influenza virus into cells mediated by H7-type HA, as well as H3 HA. Using nuclear magnetic resonance, the authors showed that the aromatic ring of TBHQ has extensive contact with the stem loop region of H7 HA [59].
Pseudovirus-based screening allowed the identification of two super-short membrane-active lipopeptides (C12-KKWK and C12-OOWO) as effective anti-IAV agents against the influenza strains A/Puerto Rico/8/34 and A/Aichi/2/68. Inhibition of virus entry occurred through the interaction of these compounds with the HA2 subunit [60].
T.C. Hung et al. conducted a screening of a number of compounds and found that quercetin (vitamin P) inhibits the entry of pseudoviruses expressing HA of the H5N1 virus into cells [49]. Studies have shown that quercetin interacts with the HA2 subunit at an early stage of influenza infection, making it a potential candidate for the development of effective, safe, and affordable natural products for the treatment and prevention of influenza A [61].
A series of oligothiophene compounds targeting the influenza virus's HA were synthesized as specific inhibitors against the H5 subtype through a series of alkylation, azidation, amination, and amidation reactions. The inhibitory activity of these compounds was tested at the cellular level against the H5N1 influenza pseudovirus. The structural analysis of these compounds showed that the size of oligothiophene compounds is very important for their level of inhibitory activity [62].
The anti-influenza activity of pyrazolo[3, 4-b]pyridinones derivatives, synthesized using an original protocol with water as a solvent, was evaluated. The protocol allowed for the synthesis of modified variants of the compounds within 1 hour. The results of the screening of the obtained compounds identified two derivatives that exhibited strong inhibitory activity against the pseudovirus A/Thailand/Kan353/2004. The speed and environmental friendliness of the synthesis of these derivatives open up new prospects in the field of drug development [63].
A series of modified C-28 pentacyclic triterpenoid saponins were synthesized through conjugation with amide derivatives, and their antiviral activity against the A/Duck/Guangdong/99 influenza virus in MDCK cells was evaluated. The study of mechanisms of action showed that these triterpenoids can strongly bind to the HA of the viral envelope, blocking the attachment of the H5N1 pseudovirus to target cells [64].
After the onset of the COVID-19 pandemic, drugs that inhibit the entry of influenza viruses were almost always studied in conjunction with drugs against SARS-CoV-2. Particularly close attention has been paid to natural compounds rather than chemically synthesized ones, as natural antiviral agents have been recognized as safe and effective.
For example, the aforementioned percolation extract of Spatholobus suberectus Dunn (SSP) is a broad-spectrum virus entry inhibitor against SARS-CoV-1/2 and other enveloped viruses. The inhibitory activity of SSP against viruses was evaluated using pseudotyped SARS-CoV-1 and 2, HIV-1ADA and HXB2, as well as H5N1.
In vivo studies have shown that even with prolonged treatment, the drug did not exhibit toxicity to the test rats compared to the control group animals. The obtained data demonstrate the potential of SSP as a candidate drug for the prevention and treatment of infections caused by enveloped viruses [37].
Griffithsin, a carbohydrate-binding protein isolated from red algae, as well as a bivalent entry inhibitor based on it (GL25E), a recombinant protein consisting of griffithsin, a 25-amino acid linker, and EK1 — a broad-spectrum coronavirus inhibitor — can effectively inhibit mono-infection with influenza A virus and SARS-CoV-2 and their co-infection by blocking HA of IAV and the spike protein of SARS-CoV-2. GL25E is more effective than griffithsin because GL25E can also interact with the HR1 domain in the S protein of SARS-CoV-2 [65].
It should be noted that the largest number of studies related to the search for antiviral drugs using pseudovirus technology is associated with the COVID-19 pandemic caused by the SARS-CoV-2 coronavirus, which has generated a real boom in virological research. A number of review publications have been published on the potential use of pseudoviruses in the study of coronavirus infections [8, 9, 65, 66]. These publications emphasize the significance of using pseudoviruses to study the interaction of SARS-CoV-2 with permissive cells, quantitatively determine neutralizing antibodies, explore new possibilities for vaccine construction, search for new antiviral drugs based on the evaluation of the activity of chemically synthesized compounds, and investigate the behavior of pseudovirus particles in the whole organism and their interaction with various organs and the immune system. The main limitations of using this technology are associated with the restricted presence of coronavirus proteins on the surface of the pseudovirus particle and the inability to use this technology to study the non-structural proteins of SARS-CoV-2. Overall, the number of publications on pseudoviruses and SARS-CoV-2 is simply enormous and requires separate consideration.
Conclusion
An analysis was conducted on the use of pseudoviruses for the development of new diagnostic, preventive, and therapeutic tools for a number of serious socially significant infectious diseases caused by RNA-containing viruses, based on data published in recent years. Currently, technologies using pseudoviruses are widely and successfully employed for research on HIV-1, hepatitis C virus, tick-borne encephalitis virus, avian influenza viruses, and SARS-CoV-2, as well as for particularly dangerous infection viruses, such as Marburg and Ebola viruses [67, 68].
The success of using pseudoviruses is determined by the fundamentally new capabilities of this technology:
- allows conducting research on the initial stage of virus entry into the cell;
- allows conducting research under conditions that ensure a high level of biosafety, especially when working with pseudoviruses that model highly pathogenic viral agents;
- principally simplifies the conduct of research and makes it possible, especially for poorly cultivable or uncultivated viruses;
- expands the experimental capabilities of researchers;
- successfully combines with modern methods of synthetic biology and bioinformatics.
The collection of presented data illustrates the fundamentally new contribution of pseudovirus technology to the search and creation of a new generation of drugs to combat serious socially significant diseases caused by RNA-containing viruses.
1 MLA style: Press release. NobelPrize.org. Nobel Prize Outreach 2025. https://www.nobelprize.org/prizes/chemistry/2024/press-release/ (data of access: 02.04.2025).
About the authors
Larisa I. Karpenko
State Research Center of Virology and Biotechnology "Vector"
Author for correspondence.
Email: lkarpenko1@ya.ru
ORCID iD: 0000-0003-4365-8809
D. Sci. (Biol.), Head, Laboratory of recombinant vaccines, leading researcher, Bioengineering department
Россия, Koltsovo, Novosibirsk regionNadezhda B. Rudometova
State Research Center of Virology and Biotechnology "Vector"
Email: nadenkaand100@mail.ru
ORCID iD: 0000-0002-1684-9071
Cand. Sci. (Biol.), senior researcher, Bioengineering department
Россия, Koltsovo, Novosibirsk regionLily F. Nizolenko
State Research Center of Virology and Biotechnology "Vector"
Email: nizolenko@inbox.ru
ORCID iD: 0000-0002-9647-4969
Cand. Sci. (Biol.), senior researcher, Bioengineering department
Россия, Koltsovo, Novosibirsk regionValery B. Loktev
State Research Center of Virology and Biotechnology "Vector"
Email: valeryloktev@gmail.com
ORCID iD: 0000-0002-0229-321X
Dr. Sci. (Biol.), Professor, chief researcher, Head, Department of molecular virology for flaviviruses and viral hepatitis
Россия, Koltsovo, Novosibirsk regionReferences
- Sagaya Jansi R., Khusro A., Agastian P., et al. Emerging paradigms of viral diseases and paramount role of natural resources as antiviral agents. Sci. Total. Environ. 2021;759: 143539. DOI: https://doi.org/10.1016/j.scitotenv.2020.143539
- Sharma K., Singh M., Sharma S.C. Revolutionizing antiviral therapeutics: in silico approaches for emerging and neglected RNA viruses. Curr. Pharm. Des. 2024;30(41):3276–90. DOI: https://doi.org/10.2174/0113816128322226240815063730
- Singh S., Kaur N., Gehlot A. Application of artificial intelligence in drug design: A review. Comput. Biol. Med. 2024;179:108810. DOI: https://doi.org/10.1016/j.compbiomed.2024.108810
- Рудометова Н.Б., Щербаков Д.Н., Рудометов А.П. и др. Модельные системы вируса иммунодефицита человека (ВИЧ-1), используемые для оценки эффективности кандидатных вакцин и лекарственных препаратов против ВИЧ-1 in vitro. Вавиловский журнал генетики и селекции. 2022;26(2): 214–21. Rudometova N.B., Shcherbakov D.N., Rudometov A.P., et al. Model systems of human immunodeficiency virus (HIV-1) for in vitro efficacy assessment of candidate vaccines and drugs against HIV-1. Vavilov Journal of Genetics and Breeding. 2022;26(2):214–21. DOI: https://doi.org/10.18699/VJGB-22-26 EDN: https://elibrary.ru/clbskg
- Welch S.R., Guerrero L.W., Chakrabarti A.K., et al. Lassa and Ebola virus inhibitors identified using minigenome and recombinant virus reporter systems. Antiviral. Res. 2016;136:9–18. DOI: https://doi.org/10.1016/j.antiviral.2016.10.007
- Chen M., Zhang X.E. Construction and applications of SARS-CoV-2 pseudoviruses: a mini review. Int. J. Biol. Sci. 2021;17(6):1574–80. DOI: https://doi.org/10.7150/ijbs.59184
- Xiang Q., Li L., Wu J., et al. Application of pseudovirus system in the development of vaccine, antiviral-drugs, and neutralizing antibodies. Microbiol. Res. 2022;258:126993. DOI: https://doi.org/10.1016/j.micres.2022.126993
- Ory D.S., Neugeboren B.A., Mulligan R.C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. USA. 1996;93(21):11400–6. DOI: https://doi.org/10.1073/pnas.93.21.11400
- Nie J., Wu X., Wang Y. Assays based on pseudotyped viruses. Adv. Exp. Med. Biol. 2023;1407:29–44. DOI: https://doi.org/10.1007/978-981-99-0113-5_2
- Wang Y., ed. Pseudotyped Viruses. Springer Singapore;2023. DOI: https://doi.org/10.1007/978-981-99-0113-5
- Montefiori D.C., Mascola J.R. Neutralizing antibodies against HIV-1: can we elicit them with vaccines and how much do we need? Curr. Opin. HIV AIDS. 2009;4(5):347–51. DOI: https://doi.org/10.1097/COH.0b013e32832f4a4d
- Li M., Gao F., Mascola J.R., et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 200;79(16):10108–25. DOI: https://doi.org/10.1128/JVI.79.16.10108-10125.2005
- Li Q., Liu Q., Huang W., et al. Current status on the development of pseudoviruses for enveloped viruses. Rev. Med. Virol. 2018;28(1):e1963. DOI: https://doi.org/10.1002/rmv.1963
- Platt E.J., Wehrly K., Kuhmann S.E., et al. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998;72(4):2855–64. DOI: https://doi.org/10.1128/JVI.72.4.2855-2864.1998
- Wei X., Decker J.M., Liu H., et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002;46(6):1896–905. DOI: https://doi.org/10.1128/AAC.46.6.1896-1905.2002
- Guo Y., Rumschlag-Booms E., Wang J., et al. Analysis of hemagglutinin-mediated entry tropism of H5N1 avian influenza. Virol. J. 2009;6:39. DOI: https://doi.org/10.1186/1743-422X-6-39
- Wang W., Nie J., Prochnow C., et al. A systematic study of the N-glycosylation sites of HIV-1 envelope protein on infectivity and antibody-mediated neutralization. Retrovirology. 2013;10:14. DOI: https://doi.org/10.1186/1742-4690-10-14
- Kretschmer M., Kadlubowska P., Hoffmann D., et al. Zikavirus prME envelope pseudotyped human immunodeficiency virus type-1 as a novel tool for glioblastoma-directed virotherapy. Cancers (Basel). 2020;12(4):1000. DOI: https://doi.org/10.3390/cancers12041000
- Wrin T., Loh T.P., Vennari J.C., et al. Adaptation to persistent growth in the H9 cell line renders a primary isolate of human immunodeficiency virus type 1 sensitive to neutralization by vaccine sera. J. Virol. 1995;69(1):39–48. DOI: https://doi.org/10.1128/JVI.69.1.39-48.1995
- Phanuphak N., Gulick R.M. HIV treatment and prevention 2019: current standards of care. Curr. Opin. HIV AIDS. 2020;15(1):4–12. DOI: https://doi.org/10.1097/COH.0000000000000588
- Arts E.J., Hazuda D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012;2(4):a007161. DOI: https://doi.org/10.1101/cshperspect.a007161
- Lobritz M.A., Ratcliff A.N., Arts E.J. HIV-1 entry, inhibitors, and resistance. Viruses. 2010;2(5):1069–105. DOI: https://doi.org/10.3390/v2051069
- Xiao T., Cai Y., Chen B. HIV-1 entry and membrane fusion inhibitors. Viruses. 2021;13(5):735. DOI: https://doi.org/10.3390/v13050735
- Solomon M., Liang C. Pseudotyped viruses for retroviruses. Adv. Exp. Med. Biol. 2023;1407:61–84. DOI: https://doi.org/10.1007/978-981-99-0113-5_4
- Westby M., van der Ryst E. CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection. Antivir. Chem. Chemother. 2005;16(6):339–54. DOI: https://doi.org/10.1177/095632020501600601
- Ding X., Zhang X., Chong H., et al. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: implications for viral entry and inhibition. J. Virol. 2017;91(18):e00831–17. DOI: https://doi.org/10.1128/JVI.00831-17
- Cocklin S., Gopi H., Querido B., et al. Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J. Virol. 2007;81(7):3645–8. DOI: https://doi.org/10.1128/JVI.01778-06
- London G.M., Mayosi B.M., Khati M. Isolation and characterization of 2'-F-RNA aptamers against whole HIV-1 subtype C envelope pseudovirus. Biochem. Biophys. Res. Commun. 2015;456(1):428–33. DOI: https://doi.org/10.1016/j.bbrc.2014.11.101
- Xu L., Zhang T., Xu X., et al. DNA triplex-based complexes display anti-HIV-1-Cell fusion activity. Nucleic Acid Ther. 2015;25(4):219–25. DOI: https://doi.org/10.1089/nat.2015.0535
- Kanizsai S., Ongrádi J., Aradi J., Nagy K. New approach for inhibition of HIV entry: modifying CD4 binding sites by thiolated pyrimidine derivatives. Pathol. Oncol. Res. 2016;22(3):617–23. DOI: https://doi.org/10.1007/s12253-016-0044-y
- Zhao Q., Ma L., Jiang S., et al. Identification of N-phenyl-N'-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339(2):213–25. DOI: https://doi.org/10.1016/j.virol.2005.06.008
- Curreli F., Belov D.S., Ramesh R.R., et al. Design, synthesis and evaluation of small molecule CD4-mimics as entry inhibitors possessing broad spectrum anti-HIV-1 activity. Bioorg. Med. Chem. 2016;24(22):5988–6003. DOI: https://doi.org/10.1016/j.bmc.2016.09.057
- Fomenko V.V., Rudometova N.B., Yarovaya O.I., et al. Synthesis and in vitro study of antiviral activity of glycyrrhizin nicotinate derivatives against HIV-1 pseudoviruses and SARS-CoV-2 viruses. Molecules. 2022;27(1):295. DOI: https://doi.org/10.3390/molecules27010295
- Фандо А.А., Фоменко В.В., Рудомётова Н.Б. и др. Модификация методики синтеза глицивира и исследование антивирусной активности полученных в ходе синтеза препаратов в отношении ENV-псевдовирусов ВИЧ-1. Химия растительного сырья. 2023;(4):387–95. Fando A.A., Fomenko V.V., Rudometova N.B., et al. Synthesis of glycivir derivatives using modification of synthesis procedure studying their antiviral activity against ENV-pseudovirouses HIV-1. Chemistry of Plant Raw Material. 2023;(4):387–95. DOI: https://doi.org/10.14258/jcprm.20230413841 EDN: https://elibrary.ru/uzjyqg
- Wang C.R., Zhou R., Ng T.B., et al. First report on isolation of methyl gallate with antioxidant, anti-HIV-1 and HIV-1 enzyme inhibitory activities from a mushroom (Pholiota adiposa). Environ. Toxicol. Pharmacol. 2014;37(2):626–37. DOI: https://doi.org/10.1016/j.etap.2014.01.023
- Mavhandu L.G., Cheng H., Bor Y.C., et al. Development of a pseudovirus assay and evaluation to screen natural products for inhibition of HIV-1 subtype C reverse transcriptase. J. Ethnopharmacol. 2020;258:112931. DOI: https://doi.org/10.1016/j.jep.2020.112931
- Liu Q., Kwan K.Y., Cao T., et al. Broad-spectrum antiviral activity of Spatholobus suberectus Dunn against SARS-CoV-2, SARS-CoV-1, H5N1, and other enveloped viruses. Phytother. Res. 2022;36(8):3232–47. DOI: https://doi.org/10.1002/ptr.7452
- Su X., Wang Q., Wen Y., et al. Protein- and peptide-based virus inactivators: inactivating viruses before their entry into cells. Front. Microbiol. 2020;11:1063. DOI: https://doi.org/10.3389/fmicb.2020.01063
- Parajuli B., Acharya K., Bach H.C., et al. Restricted HIV-1 Env glycan engagement by lectin-reengineered DAVEI protein chimera is sufficient for lytic inactivation of the virus. Biochem. J. 2018;475(5):931–57. DOI: https://doi.org/10.1042/BCJ20170662
- Беседнова Н.Н., Запорожец Т.С., Ермакова С.П., и др. Природные соединения – потенциальная основа средств профилактики и терапии гепатита С. Антибиотики и химиотерапия. 2023;68(11-12):75–90. Besednova N.N., Zaporozhets T.S., Ermakova S.P., et al. Natural compounds as potential basis for the prevention and treatment of hepatitis C. Antibiotics and Chemotherapy. 2023;68(11-12):75–90. DOI: https://doi.org/10.37489/0235-2990-2023-68-11-12-75-90 EDN: https://elibrary.ru/besoof
- Дерябин П.Г. Гепатит С: современное состояние и перспективы. Вопросы вирусологии. 2012;(S1):91–103. Deryabin P.G. Hepatitis C: current state and prospects. Problems of Virology. 2012;(S1):91–103. EDN: https://elibrary.ru/osnmkg
- Chowdhury P., Sahuc M.E., Rouillé Y., et al. Theaflavins, polyphenols of black tea, inhibit entry of hepatitis C virus in cell culture. PLoS One. 2018;13(11):e0198226. DOI: https://doi.org/10.1371/journal.pone.0198226
- Nafisi S., Roy S., Gish R., et al. Defining the possibilities: is short duration treatment of chronic hepatitis C genotype 1 with sofosbuvir-containing regimens likely to be as effective as current regimens? Expert. Rev. Anti. Infect. Ther. 2016;14(1): 41–56. DOI: https://doi.org/10.1586/14787210.2016.1114883
- Li B., Li L., Peng Z., et al. Harzianoic acids A and B, new natural scaffolds with inhibitory effects against hepatitis C virus. Bioorg. Med. Chem. 2019;27(3):560–7. DOI: https://doi.org/10.1016/j.bmc.2018.12.038
- Hung T.C., Jassey A., Liu C.H., et al. Berberine inhibits hepatitis C virus entry by targeting the viral E2 glycoprotein. Phytomedicine. 2019;53:62–9. DOI: https://doi.org/10.1016/j.phymed.2018.09.025
- Shahid M., Qadir A., Yang J., et al. An engineered microvirin variant with identical structural domains potently inhibits human immunodeficiency virus and hepatitis C virus cellular entry. Viruses. 2020;12(2):199. DOI: https://doi.org/10.3390/v12020199
- Zhang K.X., Qian X.J., Zheng W., et al. Synthesis and in vitro anti-HCV and antitumor evaluation of schisandronic acid derivatives. Med. Chem. 2021;17(9):974–82. DOI: https://doi.org/10.2174/1573406416999200818150053
- Zhang H., Zheng X., Li J., et al. Flavonoid-triazolyl hybrids as potential anti-hepatitis C virus agents: synthesis and biological evaluation. Eur. J. Med. Chem. 2021;218:113395. DOI: https://doi.org/10.1016/j.ejmech.2021.113395
- Bartosch B., Dubuisson J., Cosset F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003;197(5):633–42. DOI: https://doi.org/10.1084/jem.20021756
- Kambara H., Tani H., Mori Y., et al. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology. 2011; 412(1):211–9. DOI: https://doi.org/10.1016/j.virol.2011.01.011
- Liu H., Wu R., Yuan L., et al. Introducing a cleavable signal peptide enhances the packaging efficiency of lentiviral vectors pseudotyped with Japanese encephalitis virus envelope proteins. Virus Res. 2017;229:9–16. DOI: https://doi.org/10.1016/j.virusres.2016.12.007
- Hu H.P., Hsieh S.C., King C.C., Wang W.K. Characterization of retrovirus-based reporter viruses pseudotyped with the precursor membrane and envelope glycoproteins of four serotypes of dengue viruses. Virology. 2007;368(2):376–87. DOI: https://doi.org/10.1016/j.virol.2007.06.026
- Pan T., Peng Z., Tan L., et al. Nonsteroidal anti-inflammatory drugs potently inhibit the replication of Zika viruses by inducing the degradation of AXL. J. Virol. 2018;92(20):e01018–18. DOI: https://doi.org/10.1128/JVI.01018-18
- Zhang L., Wang X., Ming A., Tan W. Pseudotyped virus for Flaviviridae. Adv. Exp. Med. Biol. 2023;1407:313–27. DOI: https://doi.org/10.1007/978-981-99-0113-5_17
- Del Rosario J.M.M., da Costa K.A.S., Temperton N.J. Pseudotyped viruses for influenza. Adv. Exp. Med. Biol. 2023;1407:153–73. DOI: https://doi.org/10.1007/978-981-99-0113-5_8
- Huang S.W., Tai C.H., Hsu Y.M., et al. Assessing the application of a pseudovirus system for emerging SARS-CoV-2 and re-emerging avian influenza virus H5 subtypes in vaccine development. Biomed. J. 2020;43(4):375–87. DOI: https://doi.org/10.1016/j.bj.2020.06.003
- Carnell G.W., Ferrara F., Grehan K., et al. Pseudotype-based neutralization assays for influenza: a systematic analysis. Front. Immunol. 2015;6:161. DOI: https://doi.org/10.3389/fimmu.2015.00161
- Antanasijevic A., Cheng H, Wardrop D.J., et al. Inhibition of influenza H7 hemagglutinin-mediated entry. PLoS One. 2013;8(10):e76363. DOI: https://doi.org/10.1371/journal.pone.0076363
- Wu W., Wang J., Lin D., et al. Super short membrane-active lipopeptides inhibiting the entry of influenza A virus. Biochim. Biophys. Acta. 2015a;1848(10 Pt. A):2344–50. DOI: https://doi.org/10.1016/j.bbamem.2015.06.015
- Wu W., Li R., Li X., et al. Quercetin as an antiviral agent inhibits influenza A virus (IAV) entry. Viruses. 2015;8(1):6. DOI: https://doi.org/10.3390/v8010006
- Zhu Z., Yao Z., Shen X., et al. Oligothiophene compounds inhibit the membrane fusion between H5N1 avian influenza virus and the endosome of host cell. Eur. J. Med. Chem. 2017;130:185–94. DOI: https://doi.org/10.1016/j.ejmech.2017.02
- Zeng L.Y., Liu T., Yang J., et al. "On-water" facile synthesis of novel Pyrazolo[3,4-b]pyridinones possessing anti-influenza virus activity. ACS Comb. Sci. 2017;19(7):437–46. DOI: https://doi.org/10.1021/acscombsci.7b00016
- Liao Y., Chen L., Li S., et al. Structure-aided optimization of 3-O-β-chacotriosyl ursolic acid as novel H5N1 entry inhibitors with high selective index. Bioorg. Med. Chem. 2019;27(18):4048–58. DOI: https://doi.org/10.1016/j.bmc.2019.07.028
- Cao N., Cai Y., Huang X., et al. Inhibition of influenza A virus and SARS-CoV-2 infection or co-infection by griffithsin and griffithsin-based bivalent entry inhibitor. mBio. 2024;15(5):e0074124. DOI: https://doi.org/10.1128/mbio.00741-24
- Tan C., Wang N., Deng S., et al. The development and application of pseudoviruses: assessment of SARS-CoV-2 pseudoviruses. PeerJ. 2023;11:e16234. DOI: https://doi.org/10.7717/peerj.16234
- Trischitta P., Tamburello M.P., Venuti A., et al. Pseudovirus-based systems for screening natural antiviral agents: a comprehensive review. Int. J. Mol. Sci. 2024;25(10):5188. DOI: https://doi.org/10.3390/ijms25105188
- Kononova A.A., Sokolova A.S., Cheresiz S.V., et al. N-heterocyclic borneol derivatives as inhibitors of Marburg virus glycoprotein-mediated VSIV pseudotype entry. Medchemcomm. 2017;8(12):2233–7. DOI: https://doi.org/10.1039/c7md00424a
- Liu Q., Fan C., Li Q., et al. Antibody-dependent-cellular-cytotoxicity-inducing antibodies significantly affect the post-exposure treatment of Ebola virus infection. Sci. Rep. 2017;7:45552. DOI: https://doi.org/10.1038/srep45552
Supplementary files

