


Mechanisms of Toll-like receptor tolerance induced by microbial ligands
- Authors: Bulgakova I.D.1,2, Svitich O.A.1,2, Zverev V.V.1,2
-
Affiliations:
- I. Mechnikov Research Institute for Vaccines and Sera
- I.M. Sechenov First Moscow State Medical University
- Issue: Vol 99, No 6 (2022)
- Pages: 708-721
- Section: REVIEWS
- URL: https://microbiol.crie.ru/jour/article/view/1272
- DOI: https://doi.org/10.36233/0372-9311-323
- ID: 1272

Cite item

Abstract
Some microorganisms can develop tolerance. On the one hand, it allows pathogenic microbes to escape immune surveillance, on the other hand, it provides the possibility to microbiota representatives to colonize different biotopes and build a symbiotic relationship with the host. Complex regulatory interactions between innate and adaptive immune systems as well as stimulation by antigens help microbes control and maintain immunological tolerance. An important role in this process belongs to innate immune cells, which recognize microbial components through pattern-recognition receptors. Toll-like receptors (TLRs) represent the main class of these receptors. Despite the universality of the activated signaling pathways, different cellular responses are induced by interaction of TLRs with microbiota representatives and pathogenic microbes, and they vary during acute and chronic infection. The research on mechanisms underlying the development of TLR tolerance is significant, as the above receptors are involved in a wide range of infectious and noninfectious diseases; they also play an important role in development of allergic diseases, autoimmune diseases, and cancers. The knowledge of TLR tolerance mechanisms can be critically important for development of TLR ligand-based therapeutic agents for treatment and prevention of multiple diseases.
Full Text
Introduction
Immunological tolerance is a state of unresponsiveness of lymphocytes towards specific antigens. Burnet’s clonal selection theory states that lymphocyte receptors recognizing antigens are clonally distributed in the population, and the response to the binding of an antigen depends on the maturity of lymphocytes. This way the tolerance develops towards the antigens entering the body before it is immunologically mature [1–3]. Microbiota representatives start colonizing a neonate’s body and forming unique microbial communities when the immune system is still not mature [4]. Metabolites and cell components of microbiota representatives enter the bloodstream, change the functional setting of the host immune system, and regulate the sensitivity of the innate immune receptors, including TLRs [5].
Although attempts have been made to explain the mechanisms governing the changes in the sensitivity of these receptors in the context of receptor, receptor-signaling, and epigenetic theories [6], they have failed to offer a concept, which would incorporate all the findings obtained from the studies of TLR tolerance. In-depth exploration of different types of TLRs, their ligands, and activated intracellular signaling pathways as well as the analysis of genes and characteristics of the epigenetic regulation are essential for understanding the mechanisms of TLR tolerance development.
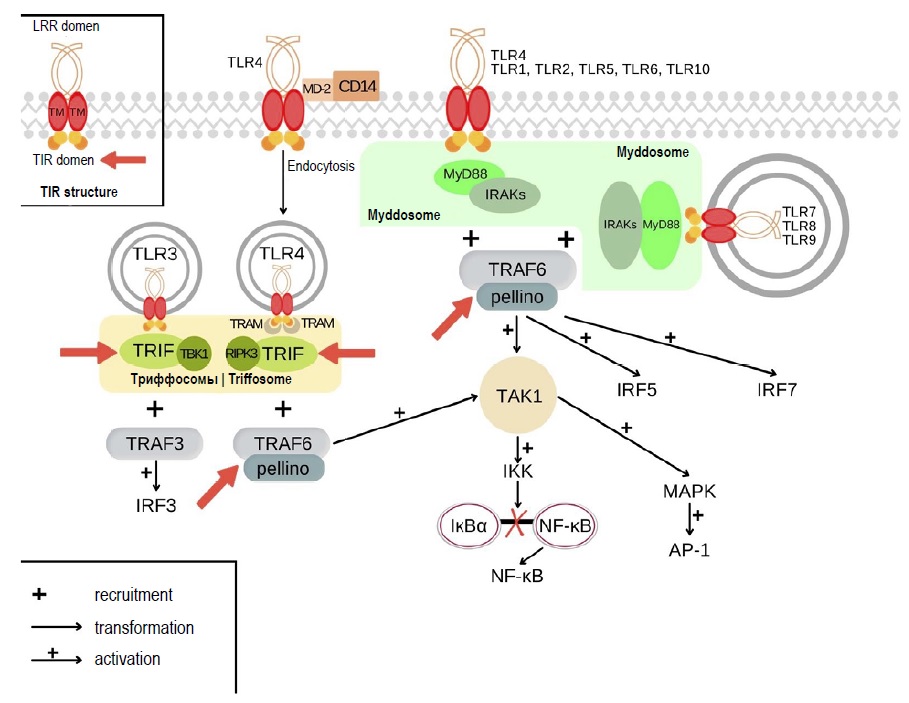
TLRs are innate immune receptors, which can recognize PAMPs (pathogen-associated molecular patterns) and DAMPs (damage-associated molecular patterns). Humans have 10 types of TLRs: TLR1, TLR2, TLR4, TLR5, TLR6, TLR10 reside on the cytoplasmic membrane, while TLR3, TLR7, TLR8, and TLR9 reside on endolysosomal membrane [7, 8]. Following ligand binding, TLRs form homodimers or heterodimers and recruit signaling components, which include adaptor proteins, kinases, and transcription factors. The schematic diagram of TLR signaling pathways is shown in Fig. 1 [7, 9, 10]. Each signaling pathway participates in formation of supramolecular organizing centers (SMOCs). All TLRs, except for TLR3, participate in activation of a MyD88-dependent pathway, in which a SMOC is represented by a myddosome consisting of the cytosolic adaptor protein MyD88. The involvement of this signaling pathway results in activation of AP-1, NF-κB, and IRF5 transcription factors, followed by the induced expression of antimicrobial factors and inflammatory mediators, and by the regulation of apoptosis [7, 11]. Stimulation of TLR3 and TLR4 receptors triggers activation of a MyD88-independent signaling pathway with the key adaptor protein TRIF, which participates in formation of another SMOC – a triffosome. The involvement of this pathway causes activation of the IRF3 transcription factor responsible for the expression of type I interferons. In addition, there are other effects associated with TLRs, which are present in non-immune cells [7, 11, 12].

Fig. 1. TLR signaling pathways, myddosome and triffosome assembly. Red arrows show the possible mechanisms underlying tolerance development at the SMOC level.
Based on the above, we can conclude that TLRs have multiple functions, and there are specific mechanisms responsible for the "switchover" of cellular responses in different conditions. With few exceptions, the type of a ligand is of no significance. Among TLR ligands, there are exogenous and endogenous ligands. Exogenous ligands (PAMPs) are represented by lipopolysaccharide (LPS), peptidoglycan, teichoic and lipoteichoic acids, flagellin, zymosan, viral DNAs and RNAs, nucleoside analogs, etc. Some types of receptors are mainly sensitive to components of specific microbes: gram-positive (TLR1, TLR2, TLR6) and gram-negative (TLR4, TLR5) bacteria, viruses (TLR3, TLR7, TLR8, TLR9), protozoa, and fungi (TLR1, TLR2, TLR6). Different DAMPs can act as endogenous ligands, for example, heat-shock proteins (Hsp60, Hsp70, Hsp96), defensins, fibrinogen [13, 14].
Despite the wide variety of ligands, it is still unknown why in some cases there is a response to TLR stimulation, while such stimulation induces no response in other cases, what mechanisms regulate these processes, and whether the nature of the ligands, their quantities, TLR stimulation frequency, type of cells and other factors are important. The answers to these questions can be received by the in-depth studying of such a phenomenon as TLR tolerance.
TLR tolerance
TLR tolerance is unresponsiveness or low responsiveness to the TLR stimulation. Earlier studies using in vivo models led to the erroneous assumption that fever could be an indicator of TLR sensitivity to endotoxins; therefore, the mechanism underlying the development of TLR tolerance was seen as desensitization of these receptors. When it was discovered that specific intracellular signaling mechanisms were activated by TLR ligation, it became clear that tolerance developed due to the altered response to the stimulation, rather than due to the desensitization of the receptors. It was first demonstrated using TLR4 and endotoxin as a ligand [15]. The phenomenon was defined as LPS-induced tolerance, though it is not the only ligand capable of inducing TLR tolerance; therefore, further on, we will use the term "induced tolerance". The negative-feedback regulation causes a decrease in released proinflammatory cytokines, thus preventing the risk of uncontrolled or inadequate inflammatory responses and subsequent tissue damage, which can be caused by prolonged or repeated exposure to TLR ligands [15, 16].
Thus, the receptor theory of TLR tolerance has been expanded into the receptor-signaling theory. However, later studies have discovered gene-specific regulatory mechanisms involved in modification of TLR-induced cellular responses. The experiment has shown that prolonged exposure to LPS resulted in various changes in chromatin; as a result, two classes of genes, tolerizeable and non-tolerizeable, have been identified. This discovery gave birth to a new epigenetic theory of TLR tolerance development [15]. Thus, induced TLR tolerance is a fundamental shift of transcription from a pro-inflammatory to an anti-inflammatory response, while retaining the protective function of innate immunity in the context of chronic or ongoing infection; however, this process is not universal and selective due to a wide variety of patterns of cytokine expression. The schematic diagram of induced TLR tolerance is shown in Fig. 2.

Fig. 2. Simplified schematic diagram of induced and cross TLR tolerance.
The basic principle is that the stronger the initial gene activation is, the more effective the induced tolerance will be [15–17]. It is assumed that the cell response to TLR stimulation follows the "all or nothing" pattern; however, the threshold value of the signal, which is required to have components of signaling pathways involved, is not constant and is regulated by SMOCs [7, 11, 18]. For example, the analysis of individual cells TLR stimulation with different doses of PAMPs has demonstrated that the rate of induced translocation of NF-κB to the nucleus does not depend on ligand levels. Any increase in the dose will only change the percentage of cells, which permit NF-κB translocation [19].
Note that TLR tolerance is reversible. The changed response to the repeated stimulation of cells is retained due to the modification of chromatin in tolerized genes; however, these changes can be reversed after a while or in response to competing signals [15, 20]. The reversibility of induced tolerance has been studied in vivo, using LPS-tolerized mouse macrophages. After treatment with granulocyte macrophage colony-stimulating factor or IFN-γ, followed by the injection of the second dose of LPS, the mice demonstrated a partial recovery of the tumor necrosis factor-α (TNF-α) and interleukin (IL)-10, but the levels of the control group were not reached [21].
The studies of components of signaling pathways associated with different TLRs led to the discovery of cross-tolerance when the initial exposure of cells to one TLR ligand induces tolerance toward the effect of ligands on other types of TLRs. Such tolerance can occur only between the receptors, activation of which results in the involvement of the same initial components of signaling pathways [6, 22–24]. The schematic diagram of TLR cross-tolerance is shown in Fig. 2.
When the TRL ligation causes the involvement of different adaptor proteins, but eventually the same transcription factors are activated, the priming effect or enhancement of the cellular response, which is opposite to tolerance, can be observed. This phenomenon correlates with the concept suggesting that SMOCs can regulate the threshold value of the signal [6, 23, 24].
Cross-tolerance may not be as effective as the tolerance induced by the repeated stimulation of one type of TLRs, i.e. autotolerance. For example, the cells initially treated with macrophage-activating lipopeptide 2 – TLR2 ligand (MALP-2) do not respond to the subsequent stimulation with LPS (TLR4 ligand), while the cells initially treated with LPS do not respond to the stimulation with lipoteichoic acid (TLR2 ligand) or flagellin (TLR5 ligand). However, the initial treatment of cells with lipoteichoic acid, LPS, and CpG (TLR9 ligand) resulted in induced autotolerance of each TLR toward these ligands, though cross-tolerance was induced by lipoteichoic acid and LPS rather than by CpG. These findings demonstrate that TLR cross-tolerance is implemented through different mechanisms [23].
Such selectiveness of cross-tolerance helps preserve the adequate immune response to certain pathogens, which is especially important in the context of the antiviral immune response. For example, the TNF-α production is inhibited in the macrophages that are tolerant to all tested TLR ligands; however, the production of other cytokines such as IL-6 is inhibited in the cells that are tolerant due to TLR4 and TLR3 ligation, while the production of interferon-β1 is inhibited in the cells tolerized by TLR4 and TLR2 ligands. The absent repression of the genes responsible for production of interferon-β1 in the cells tolerized by TLR3 ligand can imply the importance of interferons in the antiviral immune response. Likewise, the production of CXCL9 and CXCL10 chemokines by macrophages with tolerized TLR3 correlates with the role these factors play in migration of CD8+ T cells to the sites of viral infection. The repeated stimulation of TLR9 or TLR2-tolerant cells induces IL-10 gene expression at the levels comparable with the stimulation of naïve macrophages, while the initial treatment of cells with TLR4 and TLR3 ligands results in decreased production of IL-10 [16].
As mentioned previously, receptor-signaling and epigenetic theories are the main theories explaining the phenomenon of TLR tolerance. However, due to new findings about mechanisms underlying TLR tolerance, this phenomenon goes beyond the confines of one concept; therefore, some of the currently known specific mechanisms will be discussed below.
Regulatory mechanisms of TLR tolerance development during delivery of the ligand to the receptor
Interaction of the ligand with some TLRs involves additional components. Consequently, the tolerance development depends both on the shortage of these components and on their excess amount. The mechanisms involved in the tolerance development are going to be different. In the first case, the components of signaling pathways are not activated due to the disruption in the formation of the receptor complex, while induced tolerance develops in the second case. As previously described, the induced TLR tolerance is directly associated with the prior activation of these receptors and super-induced status of genes [15–17]. The extracellular LPS-binding protein forms direct contacts with bacteria and alters the outer membrane to facilitate LPS extraction. LPS-binding protein transfers LPS onto the anchored and TLR4-linked CD14 co-receptor. MD-2 molecules also participate in the activation of TLR4. Thus, the active receptor complex consists of LPS, TLR4, CD14, and MD-2, where CD14 enhances the TLR4 endocytosis [25–28]. The experiment with IL-27, which stimulates expression of TLR4 and production of soluble CD14, demonstrated that IL-27 prevented the development of tolerance to LPS. However, it was also found that the elevated basal expression of membrane-bound CD14 could promote CD14-mediated endocytosis and be responsible for the preservation of the tolerance to LPS in the presence of IL-27. The schematic diagram of the experiment is presented in Fig. 3 [29].

Fig. 3. IL-27 enhances the expression of soluble CD14 (sCD14), resulting in the completely recovered TLR4 sensitivity to LPS in the cells with a low expression level of membrane CD14 (on the left) and causing the increased production of TNF-α. The cells with high levels of membrane CD14 expression (on the right) retain a state of LPS-induced tolerance, despite the presence of IL-27, which is manifested in low production levels of TNF-α.
Intestinal epithelial cells provide another example. These cells have apical, basal, and lateral surfaces and express TLRs. While the basolateral TLR9 stimulation mobilizes the inflammatory cascade, the apical TLR9 stimulation delivers negative signals that curtail inflammatory responses induced by the basolateral stimulation by other TLRs (Fig. 4). On the one hand, such stimulation supports homeostasis; on the other hand, it can be one of the mechanisms of TLR tolerance to representatives of the intestinal microbiota [30, 31].

Fig. 4. Mechanism of suppression of basolateral TLR9 stimulation by apical delivery of TLR9 signals.
Regulatory mechanisms of TLR tolerance development during ligand and receptor interaction
Such mechanisms can include interaction of TLRs with antagonists and disruption of the receptor complex formation. Here, not only the type of the receptor, but also the nature of the ligand is important. This regulatory mechanism is of special significance for TLR4. Firstly, this receptor participates both in the MyD88-dependent and in the MyD88-independent transduction pathway, producing different effects following their activation. Secondly, as mentioned above, MD-2 molecules and CD14 co-receptor are required for activation of the MyD88-independent pathway through TLR4 [32, 33]. There are microbial TLR4 antagonists that can selectively block the activation of the surface TLR4 due to long-chain aliphatic fatty acids, which are located in the MD-2 binding cavity. Such ligands include LPS from the photosynthetic bacteria Rhodobacter sphaeroides, which have been isolated from deep lakes, as well as LPS from cyanobacteria. After administration of these TLR4 antagonists or their synthetic analogs, the subsequent TLR4 ligation with LPS from E. coli O111:B4 did not result in activation of intracellular signaling pathways. Thus, such TLR4 antagonists decrease dimerization of the TLR4–MD-2–agonist complexes, thus preventing TLR4 activation; they also inhibit downstream intracellular signaling pathways [34].
The similar mechanism of tolerance development is available for TLR2. The staphylococcal superantigen-like protein 3 (SSL3) surrounds the entrance to the lipopeptide binding pocket in the TLR2 ectodomain, preventing the access of agonists to the receptor cavity, and disrupts recruitment of the downstream adaptor protein due to limited conformational changes, which take place after TLR2 interaction with the lipopeptide [35].
Another example is based on the findings of the studies focusing on mechanisms of tolerance to microbiota representatives. Commensal bacterial LPS often has a modified structure, which affects its recognition by TLRs. Some species of Bacteroides usually contain penta-acylated and monophosphoryl lipid A structures as a dominant component of LPS. These structures are characterized by poor activation of TLR4-dependent inflammatory responses. Following the concept that minimization of TLR4 signaling is an important aspect of commensalism, most of the Bacteroides representatives present in the human intestine encode the LpxF enzyme that is responsible for production of monophosphoryl lipid A [36, 37].
The studies of multiple sclerosis have shown that the levels of L654 (TLR2 ligand), the source of which are microbiota representatives, were significantly low in such patients. During the further studies, the assumption was made that products from microbiota, such as L654, could enter the blood circulation system and cause a state of relative TLR tolerance. Thus, when the circulating levels of microbiota components are not sufficient, the normal induction of TLR tolerance can be insufficient; as a result, the TLR2 activation threshold is decreased, and larger numbers of cells start producing proinflammatory cytokines. This can promote development of autoinflammatory diseases such as multiple sclerosis [38, 39].
Regulatory mechanisms of TLR tolerance development during SMOC formation
Apparently, the events that take place in the cell after TLR ligation are not as easy to explain as it was thought previously. Each signaling pathway is involved in the formation of SMOCs. It is assumed that these structures play an important role in amplification of the signal so that it could reach the threshold value and in the specificity of cellular responses. The transduction of signals from TLRs involves two types of SMOCs – the myddosome (with the MyD88 protein as a core component) and the triffosome (with the TRIF protein constituting the core) [7, 10, 11, 17, 40].
The assembly of myddosomes following the activation of TLR2, TLR4, and TLR9 involves the adaptor protein MAL, which is responsible for interaction with the MyD88 protein, resulting in recruitment of the IRAK family kinases and TRAF6 to the myddosome. The signal transduction along the MyD88-dependent pathway through the other TLRs and the assembly of myddosomes follow the same pattern, but without participation of the adaptor protein MAL, though the intracellular events that take place after the ligation of some receptors have not been studied sufficiently [7, 9, 11].
The triffosome is assembled after TLR3 is activated, recruiting the adaptor protein TRIF, interacting with the TRAF3 ubiquitin ligase, and activating the TBK1 kinase [9, 11, 34]. The TLR4 activation does not always result in triffosome formation. Apparently, this pathway requires TLR4 endocytosis for its implementation. This process may involve the active receptor complex consisting of TLR4, CD14, and MD-2, where CD14 is responsible for TLR4 endocytosis. The adaptor protein TRAM interacts with TRIF, and this interaction results in TRAF6 recruitment to the triffosome [7, 9, 11, 27–29, 34].
Some pathogenic microbes use protein-based virulence factors to disrupt the activation of intracellular signaling pathways by affecting the SMOC components. The targets described below are shown in Fig. 1. For example, an increasing number of bacteria and viruses encode TIR-domain-containing proteins, which interact unproductively with myddosome components. Mechanisms underlying these non-productive interactions are still not clearly identified, but mutant strains deficient in proteins containing the TIR-domain can cause strong inflammatory responses and are non-virulent [41–43].
The additional strategy used by pathogenic microbes to inhibit TLR signals involves protease encoding. For example, hepatitis C virus and coxsackieviruses encode TRIF-cleaving proteases, causing inhibition of signals from TLR3 [7].
Different TLRs engage different combinations of adaptor molecules; therefore, the response to specific TLR agonists captures the combination of enzymes and substrates, which are recruited onto the specific receptor/adaptor complex. Some pathogenic bacteria and viruses can simultaneously affect several substrates, thus making it difficult to identify individual effects and assess their role in development of TLR tolerance [7].
Such mechanisms can be thoroughly studied using models with certain genes being knocked out. As for triffosome, TRIF-knockout mice can be taken as an illustrative example. Such deficiency affects both TLR3 and TLR4-mediated expression of IFN-β and activation of IRF-3. The example of the myddosome-level induced tolerance are proteins acting as a ubiquitin ligase (TRAF6) as well as E3 ubiquitin ligases pellino-1 and 2, which can overlap the activities of TRAF6. The cells lacking all three of these enzymes are defective in terms of IL-1 production. The cells lacking TRAF6 alone are not defective for these responses. In addition, TRAF6 mutants that lack enzymatic activity retain the ability to mediate rapid myddosome-directed transcriptional responses, but these responses cannot be sustained [44, 45].
Thus, the specific mechanisms underlying the tolerance development during SMOC formation have been insufficiently studied; however, myddosomes and triffosomes can be potential targets in development of induced tolerance so that pathogenic microbes would be able to evade the immune response.
Regulatory mechanisms of TLR tolerance development by regulation of transcription factors and gene repression
Multiple studies support the importance of NF-κB in pro-inflammatory gene induction. TLR tolerance depends primarily on NF-κB autoregulation, while the type of a ligand is not important. The genes repressed during tolerance are mainly associated with the NF-κB-dependent transcription, while IRF and B-ZIP motifs are amply represented in gene promoters, which are superinduced in tolerant cells. The NF-κB transcription factor plays a key role as an activator of pro-inflammatory genes of all TLRs and induction of their tolerance [15, 17, 46, 47]. For example, hepatitis C virus proteins can suppress NF-kB nuclear translocation in dendritic cells [48]. Tolerance can also be induced by regulation of other transcription factors [7, 49].
As mentioned previously, during the development of induced tolerance, the levels of cytokines and chemokines decreased nonuniformly, even though the expression levels of these genes were regulated by the same intracellular mechanisms. Therefore, it was assumed that only some of the genes were able to be repressed following the induced TLR tolerance. This concept is supported by the results of the transcriptome analysis, which were obtained after the interaction of TLR4 with the classic ligand – LPS. Two classes of genes have been identified: tolerizeable genes, which were repressed during ligation, and non-tolerizeable genes, which were not repressed [15, 50, 51]. The functional classification of LPS-inducible genes has shown that proinflammatory factors mainly belong to the class of tolerizeable genes, while genes encoding antimicrobial factors, including antimicrobial peptides and scavenger receptors, fall into the class of non-tolerizeable genes [15].
Regulatory mechanisms of TLR tolerance development through non-coding RNA and histone modification
The recent studies have found that non-coding RNAs (ncRNAs), such as small non-coding RNA molecules or microRNAs (miRs) and long non-coding RNAs (lncRNAs), can modulate an immune response. Most miRs are activated or inhibited after TLRs interact with some ligands. Such miRs participate in regulation of signaling pathways, having an effect on MyD88, TRIF, IRAKs, and TRAF6 as well as IRF3, NF-kB, and AP-1 [52]. In addition, secreted miRs can penetrate microbial cells, thus causing changes in the microbiota composition and immunological tolerance [53]. It is known that the lncRNA expression increases or decreases following the interaction of ligands with TLRs. Genes encoding lncRNAs often rank among the most dynamically regulated genes in TLR-activated cells and act as positive or negative regulators of this activation [54].
The regulatory mechanisms of TLR tolerance development through histone modification also cause changes in the gene expression during tolerance to LPS [55, 56]. Studies of some LPS-sensitive genes imply that gene promoters are also dynamically regulated, leading to tolerance. For example, transcription-associated histone H3K4 trimethylation is induced at promoters in response to LPS stimulation. However, during tolerance, H3K4 trimethylation is no longer activated at the promoters of tolerized genes such as genes responsible for production of IL-6; rather, it is induced only at the promoters of non-tolerized genes. Treatment with pargyline, an inhibitor of H3K4-demethylase, can result in H3K4 methylation at the IL-6 gene promoter and decrease the suppression of IL-6 during tolerance [55].
"Trained" innate immunity and induced TLR tolerance
As noted above, induced tolerance can be reversible; however, some cells can hold a memory, thus suggesting that the processes of induced tolerance development can have similarities with the phenomenon of "trained" innate immunity. The indirect proof of this is offered by the data obtained during studies of transcriptomic profiles of macrophages that recovered from their tolerant state. The recovery from tolerance led to the activation of their hybrid form, in which they retained characteristics of M1 and M2 [7, 57–59].
Mechanisms of tolerance and trained innate immunity are similar in that they are apparently regulated at the level of cytokine genes, which is indirectly confirmed by noticeable modifications of histones. However, the connection between these two phenomena, which are responsible for opposite effects, remains unclear. Another question is what specific intracellular events are associated with trained innate immunity and what events are associated with induced tolerance. Further research is required to explore the causes, conditions, metabolic changes in the cell, and regulatory mechanisms involved in these processes [15].
Conclusion
Recent studies have significantly broadened the knowledge of molecular mechanisms associated with TLR signaling pathways, though these receptors are still a new area of research offering big promises for clinical use. At present, quite a large number of products targeting TLRs or downstream components of signaling pathways are being evaluated through clinical trials [38, 60–63].
However, the mechanisms of induced tolerance and cross-tolerance as well as the phenomenon of trained innate immunity have been studied insufficiently. There is a high risk of adverse effects, which can develop over time or under certain conditions. The present-day studies of TLR tolerance cover only some aspects of its regulation. Further research is required to gain a deeper insight into the above process.
About the authors
Irina D. Bulgakova
I. Mechnikov Research Institute for Vaccines and Sera; I.M. Sechenov First Moscow State Medical University
Author for correspondence.
Email: bulgakova_i_d@staff.sechenov.ru
ORCID iD: 0000-0002-2629-9616
junior researcher, Laboratory of molecular immunology, postgraduate and assistant of the Microbiology, virology and immunology department
Russian Federation, Moscow; MoscowOxana A. Svitich
I. Mechnikov Research Institute for Vaccines and Sera; I.M. Sechenov First Moscow State Medical University
Email: svitich_o_a@staff.sechenov.ru
ORCID iD: 0000-0003-1757-8389
D. Sci. (Med.), Corresponding member of RAS, Head, Professor of Microbiology, virology and immunology department
Russian Federation, Moscow; MoscowVitaly V. Zverev
I. Mechnikov Research Institute for Vaccines and Sera; I.M. Sechenov First Moscow State Medical University
Email: zverev_v_v@staff.sechenov.ru
ORCID iD: 0000-0002-0017-1892
D. Sci. (Med.), Academician of RAS, Scientific head, Professor, Head, Microbiology, virology and immunology department
Russian Federation, Moscow; MoscowReferences
- Kozlov V.A. Immune paradigm and immunosuppressive dominance in the pathogenesis of major diseases of the modern man. Byulleten' Sibirskoy meditsiny. 2019; 18(1): 7–17. https://doi.org/10.20538/1682-0363-2019-1-7-17 (in Russian)
- Burnett D.L., Reed J.H., Christ D., Goodnow C.C. Clonal redemption and clonal anergy as mechanisms to balance B cell tolerance and immunity. Immunol. Rev. 2019; 292(1): 61–75. https://doi.org/10.1111/imr.12808
- Hodgkin P.D. Modifying clonal selection theory with a probabilistic cell. Immunol. Rev. 2018; 285(1): 249–62. https://doi.org/10.1111/imr.12695
- Dzhafarova K.A., Dzhafarov E.M. Role of the microbiota in immunity and inflammation. Biomeditsina (Baku). 2020; 18(3): 4–9. https://doi.org/10.24411/1815-3917-2020-11811 (in Russian)
- Belkaid Y., Harrison O.J. Homeostatic immunity and the microbiota. Immunity. 2017; 46(4): 562–76. https://doi.org/10.1016/j.immuni.2017.04.008
- Nikolaeva A.M., Maksimchik P.V., Pashchenkov M.V. A comparative characterization of macrophages tolerant to NOD1 and TLR4 receptor agonists. Immunologiya. 2021; 42(2): 102–11. https://doi.org/10.33029/0206-4952-2021-42-2-102-111 (in Russian)
- Fitzgerald K.A., Kagan J.C. Toll-like receptors and the control of immunity. Cell. 2020; 180(6): 1044–66. https://doi.org/10.1016/j.cell.2020.02.041
- Mukherjee S., Huda S., Sinha Babu S.P. Toll-like receptor polymorphism in host immune response to infectious diseases: A review. Scand. J. Immunol. 2019; 90(1): e12771. https://doi.org/10.1111/sji.12771
- Kawai T., Akira S. TLR signaling. Semin. Immunol. 2007; 19(1): 24–32. https://doi.org/10.1016/j.smim.2006.12.004
- Gay N.J., Symmons M.F., Gangloff M., Bryant C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014; 14(8): 546–58. https://doi.org/10.1038/nri3713
- Kagan J.C., Magupalli V.G., Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 2014; 14(12): 821–6. https://doi.org/10.1038/nri3757
- Snyder M., Snyder G.A. Cobbling together the myddosome. Structure. 2020; 28(6): 598–600. https://doi.org/10.1016/j.str.2020.05.006
- Vidya M.K., Kumar V.G., Sejian V., Bagath M., Krishnan G., Bhatta R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018; 37(1): 20–36. https://doi.org/10.1080/08830185.2017.1380200
- Azbukina N.V., Astakhova A.A., Goriyanov S.V., Chistyakov V.V., Sergeeva M.G. Effects of high and low molecular weight hyaluronic acids on the Omega-3 and Omega-6 fatty acid release upon activation of the Toll-like receptors in astrocytes. Biologicheskie membrany: Zhurnal membrannoy i kletochnoy biologii. 2020; 14(2): 126–33. https://doi.org/10.1134/S1990747819060035
- Foster S.L., Hargreaves D.C., Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007; 447(7147): 972–8. https://doi.org/10.1038/nature05836
- Butcher S.K., O'Carroll C.E., Wells C.A., Carmody R.J. Toll-like receptors drive specific patterns of tolerance and training on restimulation of macrophages. Front. Immunol. 2018; 9: 933. https://doi.org/10.3389/fimmu.2018.00933
- DeFelice M.M., Clark H.R., Hughey J.J., Maayan I., Kudo T., Gutschow M.V., et al. NF-κB signaling dynamics is controlled by a dose-sensing autoregulatory loop. Sci. Signal. 2019; 12(579): eaau3568. https://doi.org/10.1126/scisignal.aau3568
- Latty S.L., Sakai J., Hopkins L., Verstak B., Paramo T., Berglund N.A., et al. Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. eLife. 2018; 7: e31377. https://doi.org/10.7554/eLife.31377
- Sung M.H., Li N., Lao Q., Gottschalk R.A., Hager G.L., Fraser I.D. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-κB signaling. Sci. Signal. 2014; 7(308): ra6. https://doi.org/10.1126/scisignal.2004764
- Novakovic B., Habibi E., Wang S.Y., Arts R., Davar R., Megchelenbrink W., et al. β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. 2016; 167(5): 1354–68.e14. https://doi.org/10.1016/j.cell.2016.09.034
- Bundschuh D.S., Barsig J., Hartung T., Randow F., Döcke W.D., Volk H.D., et al. Granulocyte-macrophage colony-stimulating factor and IFN-gamma restore the systemic TNF-alpha response to endotoxin in lipopolysaccharide-desensitized mice. J. Immunol. 1997; 158(6): 2862–71.
- Dobrovolskaia M.A., Medvedev A.E., Thomas K.E., Cuesta N., Toshchakov V., Ren T., et al. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR "homotolerance" versus "heterotolerance" on NF-kappa B signaling pathway components. J. Immunol. 2003; 170(1): 508–19. https://doi.org/10.4049/jimmunol.170.1.508
- de Vos A.F., Pater J.M., van den Pangaart P.S., de Kruif M.D., van't Veer C., van der Poll T. In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-tolerance to multiple TLR ligands. J. Immunol. 2009; 183(1): 533–42. https://doi.org/10.4049/jimmunol.0802189
- Bagchi A., Herrup E.A., Warren H.S., Trigilio J., Shin H.S., Valentine C., et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J. Immunol. 2007; 178(2); 1164–71. https://doi.org/10.4049/jimmunol.178.2.1164
- Gioannini T.L., Teghanemt A., Zhang D., Coussens N.P., Dockstader W., Ramaswamy S., et al. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl Acad. Sci. USA. 2004; 101(12): 4186–91. https://doi.org/10.1073/pnas.0306906101
- Lizundia R., Sauter K.S., Taylor G., Werling D. Host species-specific usage of the TLR4-LPS receptor complex. Innate Immun. 2008; 14(4): 223–31. https://doi.org/10.1177/1753425908095957
- Park B.S., Song D.H., Kim H.M., Choi B.S., Lee H., Lee J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009; 458(7242): 1191–5. https://doi.org/10.1038/nature07830
- Zanoni I., Ostuni R., Marek L.R., Barresi S., Barbalat R., Barton G.M., et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011; 147(4): 868–80. https://doi.org/10.1016/j.cell.2011.09.051
- Petes C., Mintsopoulos V., Finnen R.L., Banfield B.W., Gee K. The effects of CD14 and IL-27 on induction of endotoxin tolerance in human monocytes and macrophages. J. Biol. Chem. 2018; 293(45): 17631–45. https://doi.org/10.1074/jbc.RA118.003501
- Lee J., Gonzales-Navajas J.M., Raz E. The "polarizing-tolerizing" mechanism of intestinal epithelium: its relevance to colonic homeostasis. Semin. Immunopathol. 2008; 30(1): 3–9. https://doi.org/10.1007/s00281-007-0099-7
- Burgueño J.F., Abreu M.T. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nature reviews. Gastroenterol. Hepatol. 2020; 17(5): 263–78. https://doi.org/10.1038/s41575-019-0261-4
- Tidswell M., Tillis W., Larosa S.P., Lynn M., Wittek A.E., Kao R., et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit. Care Med. 2010; 38(1): 72–83. https://doi.org/10.1097/CCM.0b013e3181b07b78
- Lucas K., Maes M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013; 48(1): 190–204. https://doi.org/10.1007/s12035-013-8425-7
- Di Lorenzo F., De Castro C., Silipo A., Molinaro A. Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol. Rev. 2019; 43(3): 257–72. https://doi.org/10.1093/femsre/fuz002
- Koymans K.J., Feitsma L.J., Bisschop A., Huizinga E.G., van Strijp J., de Haas C., et al. Molecular basis determining species specificity for TLR2 inhibition by staphylococcal superantigen-like protein 3 (SSL3). Vet. Res. 2018; 49(1): 115. https://doi.org/10.1186/s13567-018-0609-8
- Cullen T.W., Schofield W.B., Barry N.A., Putnam E.E., Rundell E.A., Trent M.S., et al. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science. 2015; 347(6218): 170–5. https://doi.org/10.1126/science.1260580
- Brown R.L., Larkinson M., Clarke T.B. Immunological design of commensal communities to treat intestinal infection and inflammation. PLoS Pathog. 2021; 17(1): e1009191. https://doi.org/10.1371/journal.ppat.1009191
- Anstadt E.J., Fujiwara M., Wasko N., Nichols F., Clark R.B. (2016). TLR tolerance as a treatment for central nervous system autoimmunity. J. Immunol. 2016; 197(6): 2110–8. https://doi.org/10.4049/jimmunol.1600876
- Wasko N.J., Nichols F., Clark R.B. Multiple sclerosis, the microbiome, TLR2, and the hygiene hypothesis. Autoimmun. Rev. 2020; 19(1): 102430. https://doi.org/10.1016/j.autrev.2019.102430
- Motshwene P.G., Moncrieffe M.C., Grossmann J.G., Kao C., Ayaluru M., Sandercock A.M., et al. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009; 284(37): 25404–11. https://doi.org/10.1074/jbc.M109.022392
- Bowie A., Kiss-Toth E., Symons J.A., Smith G.L., Dower S.K., O'Neill L.A. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl Acad. Sci. USA. 2000; 97(18): 10162–7. https://doi.org/10.1073/pnas.160027697
- Harte M.T., Haga I.R., Maloney G., Gray P., Reading P.C., Bartlett N.W., et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003; 197(3): 343–51. https://doi.org/10.1084/jem.20021652
- Yu H., Bruneau R.C., Brennan G., Rothenburg S. Battle royale: innate recognition of poxviruses and viral immune evasion. Biomedicines. 2021; 9(7): 765. https://doi.org/10.3390/biomedicines9070765
- Moynagh P.N. The Pellino family: IRAK E3 ligases with emerging roles in innate immune signalling. Trends Immunol. 2009; 30(1): 33–42. https://doi.org/10.1016/j.it.2008.10.001
- Strickson S., Emmerich C.H., Goh E., Zhang J., Kelsall I.R., Macartney T., et al. Roles of the TRAF6 and Pellino E3 ligases in MyD88 and RANKL signaling. Proc. Natl Acad. Sci. USA. 2017; 114(17): E3481–9. https://doi.org/10.1073/pnas.1702367114
- Carmody R.J., Ruan Q., Palmer S., Hilliard B., Chen Y.H. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007; 317(5838): 675–8. https://doi.org/10.1126/science.1142953
- Yan Q., Carmody R.J., Qu Z., Ruan Q., Jager J., Mullican S.E., et al. Nuclear factor-κB binding motifs specify Toll-like receptor-induced gene repression through an inducible repressosome. Proc. Natl Acad. Sci. USA. 2012; 109(35): 14140–5. https://doi.org/10.1073/pnas.1119842109
- Chernykh E.R., Oleynik E.A., Leplina O.Yu., Starostina N.M., Ostanin A.A. Dendritic cells in the pathogenesis of viral hepatitis C. Infektsiya i immunitet. 2019; 9(2): 239–52. https://doi.org/10.15789/2220-7619-2019-2-239-252 (in Russian)
- Song R., Gao Y., Dozmorov I., Malladi V., Saha I., McDaniel M.M., et al. IRF1 governs the differential interferon-stimulated gene responses in human monocytes and macrophages by regulating chromatin accessibility. Cell Rep. 2021; 34(12): 108891. https://doi.org/10.1016/j.celrep.2021.108891
- Mages M.J., Dietrich H., Lang R. A genome-wide analysis of LPS tolerance in macrophages. Immunobiology. 2007; 212(9-10): 723–37. https://doi.org/10.1016/j.imbio.2007.09.015
- O'Carroll C., Fagan A., Shanahan F., Carmody R.J. (Identification of a unique hybrid macrophage-polarization state following recovery from lipopolysaccharide tolerance. J. Immunol. 2014; 192(1): 427–36. https://doi.org/10.4049/jimmunol.1301722
- O'Neill L.A., Sheedy F.J., McCoy C.E. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunology. 2011; 11(3): 163–75. https://doi.org/10.1038/nri2957
- Hewel C., Kaiser J., Wierczeiko A., Linke J., Reinhardt C., Endres K., et al. Common miRNA patterns of Alzheimer's disease and Parkinson's disease and their putative impact on commensal gut microbiota. Front. Neurosci. 2019; 13: 113. https://doi.org/10.3389/fnins.2019.00113
- Carpenter S., Aiello D., Atianand M.K., Ricci E.P., Gandhi P., Hall L.L., et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013; 341(6147): 789–92. https://doi.org/10.1126/science.1240925
- Seeley J.J., Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017; 101(1): 107–19. https://doi.org/10.1189/jlb.3MR0316-118RR
- Saeed S., Quintin J., Kerstens H.H., Rao N.A., Aghajanirefah A., Matarese F., et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014; 345(6204): 1251086. https://doi.org/10.1126/science.1251086
- O'Carroll C., Fagan A., Shanahan F., Carmody R.J. Identification of a unique hybrid macrophage-polarization state following recovery from lipopolysaccharide tolerance. J. Immunol. 2014; 192(1): 427–36. https://doi.org/10.4049/jimmunol.1301722
- Fischer C., Metsger M., Bauch S., Vidal R., Böttcher M., Grote P., et al. Signals trigger state-specific transcriptional programs to support diversity and homeostasis in immune cells. Sci. Signal. 2019; 12(581): eaao5820. https://doi.org/10.1126/scisignal.aao5820
- Arts R.J., Joosten L.A., Netea M.G. Immunometabolic circuits in trained immunity. Semin. Immunol. 2016; 28(5): 425–30. https://doi.org/10.1016/j.smim.2016.09.002
- Gambuzza M.E., Sofo V., Salmeri F.M., Soraci L., Marino S., Bramanti P. Toll-like receptors in Alzheimer's disease: a therapeutic perspective. CNS Neurol. Disord. Drug Targets. 2014; 13(9): 1542–58. https://doi.org/10.2174/1871527313666140806124850
- Luchner M., Reinke S., Milicic A. TLR agonists as vaccine adjuvants targeting cancer and infectious diseases. Pharmaceutics. 2021; 13(2): 142. https://doi.org/10.3390/pharmaceutics13020142
- Anwar M.A., Shah M., Kim J., Choi S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019; 39(3): 1053–90. https://doi.org/10.1002/med.21553
- Vijay K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018; 59: 391–412. https://doi.org/10.1016/j.intimp.2018.03.002
Supplementary files

