


Разнообразие СRISPR-локусов штаммов Yersinia pseudotuberculosis и их ассоциация с факторами патогенности возбудителя
- Авторы: Перетолчина Н.П.1, Воскресенская Е.А.2, Кокорина Г.И.2, Богумильчик Е.А.2, Климов В.Т.3, Трухачев А.Л.4, Джиоев Ю.П.1, Злобин В.И.1,5
-
Учреждения:
- Иркутский государственный медицинский университет
- Санкт-Петербургский научно-исследовательский институт эпидемиологии и микробиологии имени Пастера
- Иркутский научно-исследовательский противочумный институт Сибири и Дальнего Востока
- Ростовский-на-Дону научно-исследовательский противочумный институт
- Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
- Выпуск: Том 100, № 3 (2023)
- Страницы: 166-175
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://microbiol.crie.ru/jour/article/view/2801
- DOI: https://doi.org/10.36233/0372-9311-374
- EDN: https://elibrary.ru/ornkhs
- ID: 2801

Цитировать
Полный текст

Аннотация
Актуальность. Постоянный обмен наследственной информацией с участием внехромосомных и транспозируемых элементов является одним из факторов существования гетерогенных популяций микроорганизмов. Структуры, контролирующие горизонтальный перенос генов, могут влиять на формирование генетических вариантов патогенов.
Цель работы — изучить молекулярно-генетические характеристики CRISPR-локусов штаммов Yersinia pseudotuberculosis, циркулирующих на территории России, и их связь с факторами патогенности возбудителя псевдотуберкулёза.
Материалы и методы. В работе изучены 56 штаммов Y. pseudotuberculosis, выделенных в течение 1935–2014 гг. на различных территориях России и других государств из разных источников. Выделение и амплификацию фрагментов ДНК, содержащих CRISPR-локусы, выполняли методом полимеразной цепной реакции. Секвенирование CRISPR-локусов осуществляли по методу Сэнгера. Для каждого штамма определён генотип по наличию основных факторов патогенности: хромосомных генов суперантигена ypmА/С, генов, локализованных на «островах патогенности» HPI, YAPI, генов плазмиды pYV, pVM82.
Результаты. В результате анализа CRISPR-локусов штаммов Y. pseudotuberculosis установлено, что спейсерный состав локусов различается у штаммов, отличающихся серотипом, набором факторов патогенности, регионом выделения. Наиболее вариабельны локусы YP1 и YP3, локус YP2 однороден. Состав локуса YP1 различается в группах штаммов, отличающихся по наличию плазмиды pVM82. Состав локуса YP3 зависит и от наличия плазмиды pVM82, и от «адгезивного острова патогенности» YAPI.
Заключение. На территории России циркулируют штаммы Y. pseudotuberculosis с различным набором детерминант вирулентности и спейсерным составом СRISPR-локусов. Большинство российских штаммов отличаются по данным молекулярным маркерам от штаммов из других стран. Высокая вариабельность штаммов внутри вида определяется способностью приобретать новые генетические элементы путём горизонтального переноса генов. CRISPR-Cas как система «иммунитета» бактерий против мобильных генетических элементов может участвовать в формировании патогенного потенциала штаммов Y. pseudotuberculosis.
Ключевые слова
Полный текст
Введение
Грамотрицательные бактерии вида Yersinia pseudotuberculosis относят к факультативным психрофилам, способным вызывать острое сапрозоонозное заболевание — псевдотуберкулёз [1]. Случаи псевдотуберкулёза регистрируют во многих субъектах России, но регионами с наиболее высокой заболеваемостью являются Дальневосточный, Северо-Западный и Сибирский федеральные округа. Классические методы типирования штаммов Y. pseudotuberculosis основаны на определении серотипа [2, 3]. Большинство циркулирующих на территории России штаммов относятся к серотипу О:1, преимущественно O:1b, в сибирском регионе также выявлены представители серотипа О:3, на Дальнем Востоке, кроме перечисленных, встречаются штаммы серотипа О:3, О:4 [4, 5]. Штаммы Y. pseudotuberculosis отличаются высоким разнообразием факторов, определяющих их вирулентность и тяжесть вызываемого ими заболевания. Хромосомные факторы патогенности Y. pseudotuberculosis, выявляемые методом полимеразной цепной реакции (ПЦР) включают гены суперантигена ypmA, ypmC, «острова патогенности» HPI («остров высокой патогенности») и YAPI («адгезивный остров патогенности»). На плазмидах pYV и pVM82 локализованы гены, ассоциированные с факультативным внутриклеточным паразитизмом бактерии: аппарат секреции III типа, адгезины и аппарат секреции IVB типа. Но данные методы типирования не позволяют дифференцировать близкородственные штаммы внутри одного серотипа или геноварианта с определёнными факторами патогенности. В зависимости от последующего метода генотипирования, основанного на анализе геномного полиморфизма штаммов Y. pseudotuberculosis, выявляется различное число генотипов, отличающихся серотипом, географией и источником выделения, патогенным потенциалом [6, 7]. Применение нескольких методов генотипирования позволяет получить более информативные результаты.
Постоянный обмен наследственной информацией с участием внехромосомных и транспозируемых элементов является одним из факторов существования гетерогенной популяции микроорганизмов в определённых условиях среды [1]. Некоторые кластеры генов, кодирующие детерминанты патогенности, ассоциированы с интегразами профагов или инсерционными элементами, контролирующими геномные перестройки, следовательно, системы, направленные на регуляцию горизонтального переноса генов, влияют на эволюцию бактерий. К таким структурам относится система CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats, короткие палиндромные повторы, разделённые спейсерами), способная специфически узнавать и уничтожать чужеродный генетический материал.
CRISPR-Cas-системы Y. pseudotuberculosis относятся к классу 1, типу I и подтипу F (IF), и имеют в своём составе от одного до 3 локусов (YP1, YP2 и YP3), удалённо расположенных относительно друг друга вследствие геномных перестроек. Cas-гены локализованы около одной из кассет (YP1), представленных в геноме иерсиний. Остальные локусы (YP2 и YP3) являются производными локуса YP1 и сохраняют способность функционировать в качестве CRISPR-иммунитета [8, 9]. Ранее K.A. Koskela и соавт., Т. Seecharran и соавт. обнаружили соответствие между некоторыми сиквенс-типами и спейсерным составом CRISPR-кассет штаммов Y. pseudotuberculosis [9, 10].
Но в работах, посвящённых изучению CRISPR- Cas-системе Y. pseudotuberculosis, штаммы, изолированные на территории России, представлены в относительно небольших количествах, а также отсутствует информация о месте выделения штамма и другие паспортные данные.
Целью настоящего исследования явилось изучение молекулярно-генетических характеристик CRISPR-локусов штаммов Y. pseudotuberculosis, циркулирующих на территории России, и их связи с факторами патогенности.
Материалы и методы
Изучены 56 штаммов Y. pseudotuberculosis [11], выделенные в течение 1935–2014 гг. на территориях Восточной и Западной Сибири, Дальнего Востока, Северо-Западного и других регионов России, Монголии, Украины, Беларуси, Туркменистана и Абхазии из различных источников (cм. Приложение на сайте журнала).
Штаммы отобраны из коллекций Иркутского НИПЧИ Сибири и Дальнего Востока, НИИ эпидемиологии и микробиологии им. Пастера, Ростовского-на-Дону научно-исследовательского противочумного института с учётом имеющихся паспортных данных в соответствии с полученными ранее данными о длине CRISPR-локусов [11].
Для выделения ДНК из коллекционных штаммов использовали комплект реагентов «Рибо-сорб» («AmpliSens») и «АмплиПрайм ДНК-сорб-В» (ООО «НекстБио»). ПЦР проводили на амплификаторе компании «Applied Biosystems» и на программируемом термостате «Терцик» (ЗАО «ДНК-Технология»). Для выделения и секвенирования CRISPR-локусов использовали праймеры и методику, представленные ранее [11].
Выявление хромосомных генов суперантигена ypmA/C, генов, локализованных на «островах патогенности» HPI (fyuA, irp2), YAPI (pilPQ, api74, tcp), генов плазмиды pVM82 (dotO, mucAB) проводили методом ПЦР [12].
Серотип исследуемых штаммов определяли по общепринятой методике с диагностическими сыворотками производства ФБУН НИИ эпидемиологии и микробиологии имени Пастера и методом мультиплексной ПЦР [2, 3].
Секвенирование CRISPR-локусов осуществляли по Сэнгеру на капиллярном секвенаторе «Applied Biosystems 3500» («Thermo Scientific»). Для 10 штаммов провели полногеномное секвенирование на секвенаторе MiSeq («Illumina»). Пробоподготовку и секвенирование проводили в соответствии с инструкциями производителя.
Сборку и анализ фрагментов проводили с помощью программ «Vector NTI Advance 10.3» и «BioEdit 7.2». Сборку до уровня контигов реализовывали с помощью геномного сборщика SPAdes 3.15.4 и инструмента CONTIGuator 2.7.4, осуществляющего финишную сборку на основе референсного генома [13, 14]. Поиск и расшифровку CRISPR-Сas-систем в последовательностях штаммов проводили с использованием программ «CRISPRone»1, «CRISPRCasFinder»2 и «CRISPRDetect»3 [15–17]. Гомологичные протоспейсерные последовательности в геномах бактериофагов и плазмид определяли с помощью алгоритма поиска BLASTn по базам данных GenBank, EMBL, DDBJ, PDB, RefSeq, онлайн-приложения CRISPRTarget по базам данных GenBank-Phage, RefSeq-Plasmid и Islandviewer [18, 19]. Гомологичные последовательности искали с использованием следующих критериев: наличие 3'-PAM, характерного для CRISPR-систем типа IF — GG, не более 2 несоответствий комплементарных оснований, отсутствие более 2 спаренных нуклеотидов подряд. Профаги в геномах хромосом определяли с использованием онлайн-инструмента PHASTER [20].
Анализ данных проводили в среде программирования RStudio 2022.07.1+554 с использованием языка для статистической обработки данных и работы с графикой R 4.2.2 [21]. Пермутационный дисперсионный анализ (PERMANOVA) использовали для поиска статистически значимых различий в строении локусов и их связи с различными характеристиками штаммов в составе пакета vegan 2.6-4 [22]. Кластеризацию штаммов и построение дендрограмм по наличию спейсеров проводили с использованием пакетов pvclust 2.2-0 и ComplexHeatmap 2.14.0 по методу невзвешенного попарного среднего (UPGMA) с бутстрепом 1000 [23].
Регистрационные номера последовательностей 69 нуклеотидных последовательностей CRISPR-локусов штаммов Y. pseudotuberculosis депонированы в международную базу GenBank (номера доступа: KX592595–KX592599 и OP245022–OP245086).
Результаты
В результате анализа качества и сборки сиквенсов отобраны 78 фрагментов. Показано, что исследованные штаммы Y. pseudotuberculosis содержат СRISPR-Cas-систему, включающую 1–3 локуса: YP1, YP2, YP3. Согласно структуре повторов (5’-GTT CAC TGC CGC ACA GGC AGC TTA GAA A-3') все локусы относятся к типу IF. Общее количество спейсеров составило 1311. В локусе YP1 количество спейсеров варьирует от 3 до 38 (всего 810 спейсеров), в локусе YP3 — от 1 до 44 (всего 466), локус YP2 — короткий и состоит из 1 или 2 спейсеров.
Спейсерный состав СRISPR-локусов
Для анализа спейсерного состава CRISPR-локусов была создана база данных спейсеров штаммов Y. pseudotuberculosis (№ 2020622813, зарегистрирована в Государственном реестре баз данных, 25.12.2020).
Статистически достоверно спейсерный состав CRISPR-Cas-систем различается у штаммов, отличающихся серотипом, набором факторов патогенности, выделенных на разных территориях (таблица). При изучении отдельных локусов статистически достоверно различаются составы локусов YP1 и YP3, состав локуса YP2, напротив, достаточно однороден. Спейсерный состав локуса YP1 отличается у штаммов разных серотипов и генотипов, в частности, у штаммов с различным плазмидным составом (pVM82+ или pVM82–). Состав локуса YP3, в отличие от локуса YP1, достоверно отличается у штаммов, имеющих в составе генома «адгезивный остров патогенности» YAPI и плазмиду pVM82. Статистически достоверных различий в группах штаммов, отличающихся по остальным факторам патогенности (суперантиген YPM, «остров патогенности» HPI), не выявлено.
Различия спейсерного состава CRISPR-локусов штаммов Y. pseudotuberculosis (при p < 0,05) / Differences of spacer content in CRISPR-loci of Y. pseudotuberculosis strains (p < 0,05)
Характеристика штамма Strain characteristics | Df | MS | Pseudo-F | p |
Локус YP1 | Locus YP1 | ||||
Регион выделения | Isolation place | 22 | 0,4668 | 3,9483 | 0,001 |
Серотип | Serotype | 1 | 0,6831 | 5,7779 | 0,001 |
Сочетание факторов патогенности | Combination of pathogenicity factors | 3 | 0,554 | 4,6857 | 0,001 |
pVM | 1 | 0,4659 | 3,9408 | 0,002 |
YAPI | 1 | 0,2125 | 1,7976 | 0,089 |
Другие факторы | Other factors | 10 | 0,1182 | ||
Локус YP2 | Locus YP2 | ||||
Регион выделения | Isolation place | 13 | 0,1869 | 3,3647 | 0,215 |
Серотип | Serotype | 3 | 0,0664 | 1,1944 | 0,420 |
Другие факторы | Other factors | 3 | 0,0556 | ||
Локус YP3 | Locus YP3 | ||||
Регион выделения | Isolation place | 20 | 0,4439 | 36,117 | 0,001 |
Серотип | Serotype | 3 | 0,4352 | 35,402 | 0,001 |
Сочетание факторов патогенности | Combination of pathogenicity factors | 3 | 0,5478 | 44,567 | 0,001 |
pVM | 1 | 0,1984 | 16,145 | 0,001 |
YAPI | 1 | 0,3016 | 24,537 | 0,001 |
Другие факторы | Other factors | 7 | 0,0123 | ||
Все локусы | All loci | ||||
Регион выделения | Isolation place | 27 | 0,555 | 2,6799 | 0,001 |
Серотип | Serotype | 4 | 0,3906 | 1,8858 | 0,001 |
Сочетание факторов патогенности | Combination of pathogenicity factors | 3 | 0,5049 | 2,4379 | 0,001 |
pVM | 1 | 0,5279 | 2,5487 | 0,007 |
YAPI | 1 | 0,2454 | 1,185 | 0,276 |
Другие факторы | Other factors | 19 | 0,2071 | ||
Примечание. Df — степени свободы; MS — средний квадрат; Pseudo-F — оценка критерия Фишера; p — оценка уровня значимости.
Note. Df — degree of freedom; MS — mean square; Pseudo-F — Fisher criterion; p — significance level.
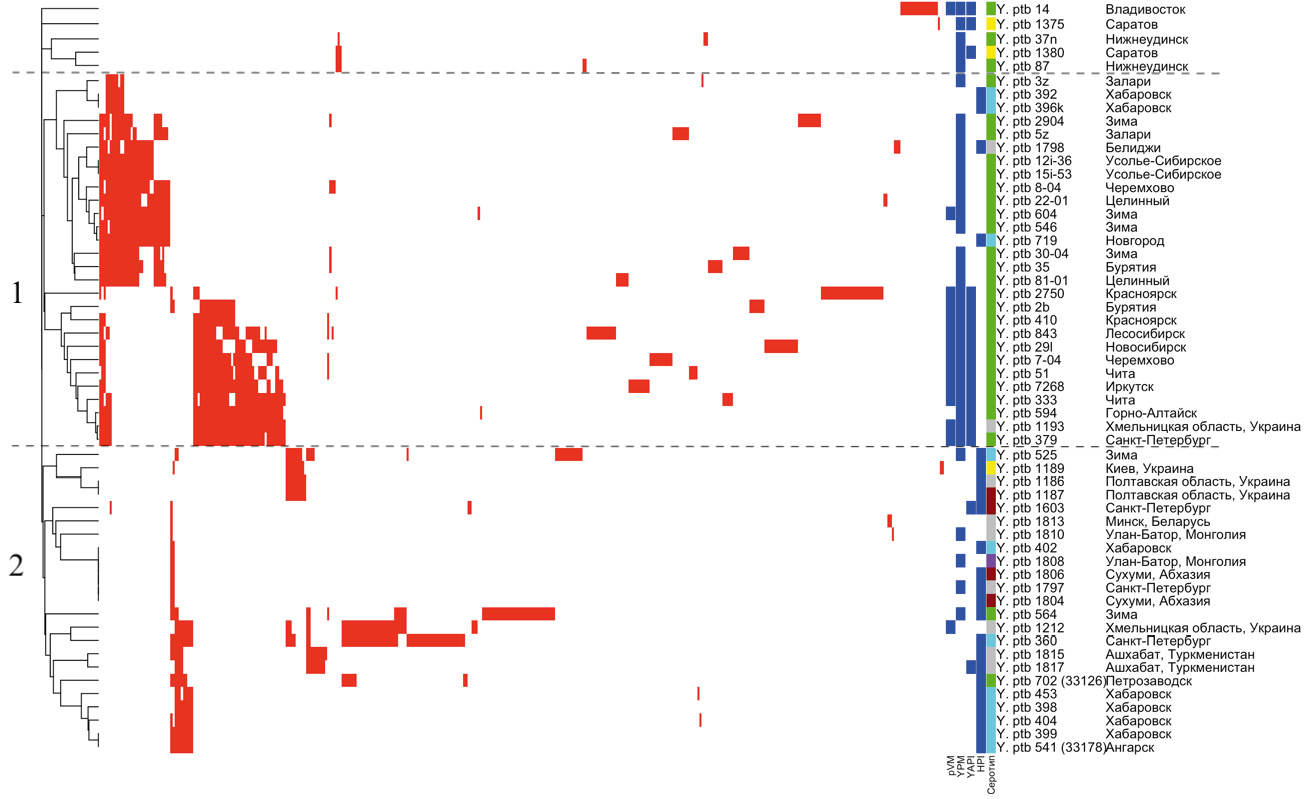
Кластерный анализ штаммов, основанный на наличии спейсеров в локусах, позволяет условно разделить штаммы на 2 большие группы (рис. 1).

Рис. 1. Кластеризация штаммов Y. pseudotuberculosis (n = 56) по наличию спейсеров в локусах YP1–YP3. Условное обозначение по цветам: наличие спейсера — красный; наличие фактора патогенности — синий; серотип 1 — тёмно-красный; серотип 1а — голубой; серотип 1b — зелёный; серотип 3 — жёлтый; серотип 4 — фиолетовый; серотип не определён — серый. / Fig. 1. Clusterization of Y. pseudotuberculosis strains (n = 56) by presence of spacer in loci YP1–YP3. Color designations: presence of spacer — red; presence of factor — blue; serotype 1 — darkred; serotype 1a — cyan; serotype 1b — green; serotype 3 — yellow; serotype 4 — violet; serotype is not defined — grey.
Кластер 1 включает 28 штаммов Y. pseudotuberculosis, выделенных преимущественно на территориях Сибирского федерального округа (86%): 3 штамма серотипа О:1а, 23 штамма серотипа О:1b (для 2 штаммов серотип не определён). Для штаммов характерно наличие суперантигена YPM (89%). При этом внутри кластера происходит разделение штаммов на 2 подкластера по наличию «адгезивного острова патогенности» YAPI и плазмиды pVM82: подкластер 1–1 (YPM+YAPI–pVM82–) включает 16 (57%) штаммов, подкластер 1–2 (YPM+YAPI+pVM82+) — 12 (43%) штаммов.
Кластер 2 состоит из 23 штаммов Y. pseudotuberculosis разных серотипов, практически в равных долях выделенных в различных регионах России (52%) и других государств (48%). Для большинства штаммов характерно наличие «острова патогенности» HPI (83%), 5 (22%) штаммов обладают суперантигеном YPM, 1 (4%) штамм — «адгезивным островом патогенности» YAPI, 1 (4%) штамм — плазмидой pVM82.
Пять (9%) из изученных 56 штаммов (14, 1375, 37n, 1380, 87) находятся вне основных кластеров. Спейсеры, представленные в данных штаммах, практически не встречаются в других СRISPR-системах.
Сравнительный анализ полученных сиквенсов с последовательностями геномов, депонированных в базах данных NCBI (Genbank и RefSeq), показал разделение штаммов на «европейскую» (группа 1) и «азиатскую» (группа 2) клады (рис. 2).

Рис. 2. Кластеризация штаммов Y. pseudotuberculosis (n = 56) по наличию спейсеров. Данные секвенирования локусов CRISPR-Cas-систем (n = 31) и сиквенсов в NCBI (n = 75). Условное обозначение по цветам: наличие спейсера — красный, наличие фактора патогенности — синий. / Fig. 2. Clusterization of Y. pseudotuberculosis strains (n = 56) by presence of spacer. CRISPR-Cas system sequenced data (n = 31) and NCBI data (n = 75). Color designations: presence of spacer — red, presence of factor — blue.
В группу 1 вошли 52 штамма, выделенных преимущественно на территории России и Японии (73%). Также встречаются штаммы (15%), выделенные в различных странах: Германии, Южной Африке, Туркменистане, Швеции, Украине и Новой Зеландии. Для большинства штаммов данной группы (87%) характерно наличие гена суперантигена ypm. Около половины штаммов (56%) несут «адгезивный остров патогенности» YAPI (в том числе ген tcp), при этом у части штаммов выявлена также плазмида pVM82 (22%).
Группа 2 включает 54 штамма, выделенных преимущественно в странах Европы (51%), за исключением 7 (13%) штаммов из других стран (Китай, азиатская часть России, Канада, Новая Зеландия, Австралия). Для большинства штаммов данной группы (59%) характерно наличие «острова патогенности» HPI. Другим преобладающим фактором патогенности является «адгезивный остров патогенности» YAPI (20%).
Таким образом, штаммы Y. pseudotuberculosis, циркулирующие на территории России и ряда других стран, нельзя чётко разделить на «азиатскую» и «европейскую» клады в зависимости от типа CRISPR-кластера, что связано с высокой вариабельностью штаммов. Вместе с тем проведённое сопоставление CRISPR-локусов с данными о наличии основных факторов патогенности позволило выявить генетические варианты штаммов Y. pseudotuberculosis, доминирующих на отдельных территориях. Можно заключить, что большинство штаммов, циркулирующих на территории России, отличается по данным молекулярным маркерам от штаммов из других стран.
Анализ протоспейсерных последовательностей в геномах фагов и плазмид
Исходя из иммунной функции CRISPR-Cas-системы, произведён поиск гомологичных последовательностей в геномах основных мобильных генетических элементов (МГЭ) — бактериофагах и плазмидах. Суммарно в CRISPR-системах обнаружено 404 уникальных спейсерных последовательности, которые встречаются в разных штаммах. Анализ протоспейсеров в геномах бактерий, бактериофагов и плазмид выявил только 101 (25%) соответствие спейсеров фрагментам геномов.
20 спейсерных последовательностей гомологичны фрагментам геномов бактериофагов Yersinia YeP3, Yersinia YeP2, Yersinia YeP1, Yersinia vB_YenM, Yersinia vB_YenS_P400 и Klebsiella 6991. Семьдесят спейсерных последовательностей гомологичны последовательностям хромосом разных видов Yersinia: Y. pseudotuberculosis, Y. pestis, Y. similis, Y. massiliensis, Y. intermedia, Y. kristensenii, Y. enterocolitica, Y. aldovae. Согласно результатам анализа геномов с помощью онлайн-инструмента PHASTER, фрагменты геномов, содержащие протоспейсерные последовательности, соответствуют полноценным профагам [20, 24]. Один и тот же спейсер может быть гомологичен фрагментам генома профагов, обнаруженных в разных штаммах. Кроме того, спейсеры, представленные в одном локусе, способны узнавать разные фрагменты генома одного и того же профага.
Семь спейсеров гомологичны последовательностям генома плазмид Y. pseudotuberculosis и Y. massiliensis, 4 спейсера гомологичны последовательности криптической плазмиды pYptb32953 размером 27,7 kb, которая встречается достаточно редко в штаммах патогенных иерсиний. Основная функция данной плазмиды — участие в конъюгации: в геноме плазмиды расположен кластер генов, кодирующих белки, необходимые для конъюгации плазмиды. Четыре спейсерных последовательности оказались гомологичны участку генома плазмиды pVM82 штаммов Y. pseudotuberculosis IP31758 и Y. pseudotuberculosis 598 (номера доступов в NCBI RefSeq: NC_009705 и NZ_CP071946), за исключением 4 нуклеотидов (рис. 3). В геноме плазмид находятся гены фагоподобной интегразы, что может свидетельствовать о приобретении кластера генов icm/dot системы секреции IVB-типа через механизмы горизонтального переноса.

Рис. 3. Протоспейсерная последовательность плазмиды Y. pseudotuberculosis IP31758 pVM82 кДa (153 кб) к спейсеру 114 локуса YP3.
Fig. 3. Protospacer sequence of plasmid pVM82 kDa (153 kb) of Y. pseudotuberculosis IP31758 to spacer 114 of locus YP3.
Обсуждение
Основная функция CRISPR-Cas-систем — это специфическое узнавание и уничтожение чужеродных генетических элементов, попавших в бактериальную клетку и представляющих потенциальную опасность для существования популяции бактерии [25, 26]. Следовательно, последовательности спейсеров будут гомологичны фрагментам геномов МГЭ, характерных для определённых территорий. Способность приобретать спейсеры в хронологическом порядке и высокая доля полиморфизмов позволяет использовать CRISPR-Cas-системы как инструмент высокоразрешающего молекулярного типирования штаммов, что было продемонстрировано на примере Streptococcus thermophilus [27], Campylobacter jejuni [26], Corynebacterium diphtheriae [28] и Y. pestis [29]. Известно, что CRISPR-локусы являются динамической структурой, где новые спейсерные последовательности присоединяются со стороны лидерной последовательности [30]. Часть спейсеров, расположенных в середине кассеты, может быть потеряна в результате делеции во время приобретения новых последовательностей МГЭ. Спейсеры, расположенные около последнего полиморфного повтора, как правило, сохраняются [31].
Так, типирование штаммов по наличию спейсеров, расположенных в конце локусов YP1 и YP3, позволяет условно разделить штаммы, различающиеся набором детерминант вирулентности, на два кластера: «европейский» и «азиатский». Из исследованных 56 штаммов 35 выделены на территориях Дальнего Востока, Восточной и Западной Сибири. При этом происходит разделение на две подгруппы в зависимости от присутствия плазмиды pVM82 (pVM82+ и pVM82–), детерминирующей аппарат секреции IVB-типа и, наряду с другими факторами патогенности, опосредующей развитие генерализованных форм псевдотуберкулёза. Штаммы, выделенные в Японии и Корее, также способные вызывать комплекс проявлений системной инфекции, названной дальневосточной скарлатиноподобной лихорадкой, имеют схожий паттерн распределения CRISPR-кассет.
Согласно результатам анализа PERMANOVA, состав локусов YP1 и YP3 зависит от наличия у штаммов таких факторов патогенности, как плазмида pVM82 и «адгезивный остров патогенности» YAPI. Полученные результаты согласуются с ранее описанными отличиями длины локуса YP3: штаммы, имеющие данные основные детерминанты патогенности и вызывающие дальневосточную скарлатиноподобную лихорадку, отличаются более длинным локусом YP3 по сравнению со штаммами, у которых pVM82 и YAPI не обнаружены [11]. K.A. Koskela и соавт. показали, что некоторые сиквенс-типы штаммов коррелируют со спейсерным составом CRISPR-кассет [9]. Ранее проведённый Т. Seecharran и соавт. филогенетический анализ штаммов Y. pseudotuberculosis выявил высокую степень разнообразия CRISPR-локусов среди штаммов, выделенных на азиатском континенте [10]. Пангеномный анализ штаммов Y. pseudotuberculosis не показал значимых соответствий в паттернах CRISPR-кассет и уникального геномного профиля штаммов [10]. Наиболее выраженные различия кластеров наблюдаются в вариабельном геноме, гены которого часто теряются или приобретаются путём горизонтального переноса.
Высокая вариабельность штаммов внутри вида определяется способностью приобретать новые генетические элементы (геномные острова) путём горизонтального переноса генов с помощью плазмид, бактериофагов, интегронов и транспозонов [32]. CRISPR-Cas как система «иммунитета» бактерий против МГЭ может участвовать в формировании генотипа штаммов Y. pseudotuberculosis, определяющего клинические проявления псевдотуберкулёза.
Анализ протоспейсеров в геномах бактерий, бактериофагов и плазмид выявил соответствие спейсеров участку генома плазмиды pVM82 штаммов Y. pseudotuberculosis IP31758 и Y. pseudotuberculosis 598. Возможно, приобретение данных спейсеров может быть причиной отсутствия плазмиды pVM82 в штаммах, относящихся к «европейскому» кластеру. Спейсер, способный узнавать участок плазмиды, обнаруживается в локусе YP3 штамма Y. pseudotuberculosis 2750 серотипа О:1b, содержащего плазмиду pVM82 и кластер генов icm/dot. Вероятно, большое количество мутаций в протоспейсерной последовательности препятствует CRISPR-интерференции или данная последовательность может присутствовать в непредставленном в базах данных геноме бактериофага. Кроме того, приобретение спейсеров к собственным последовательностям может служить триггером для возникновения новых мутаций и экспрессии нового фенотипа, проявляющегося в изменении вирулентных свойств микроорганизма [25]. Согласно другой гипотезе спейсеры, узнающие собственные последовательности, являются механизмом селекции штаммов, не содержащих данный фактор [33]. Таким образом, CRISPR-Cas-система может являться одним из факторов, определяющих циркуляцию разнообразных штаммов на одной территории. В сочетании с классическими методами идентификации и типирования возбудителя псевдотуберкулёза анализ CRISPR-локусов может быть использован для филогенетических исследований, мониторинга циркуляции штаммов Y. pseudotuberculosis на территориях, эпидемиологического расследования групповых заболеваний.
Метод секвенирования ПЦР-фрагментов по Сэнгеру имеет ряд ограничений, в частности невозможность секвенирования фрагментов длиной более 1000 п.н. Развитие методов секвенирования и накопление данных о бактериальных геномах в будущем позволит провести анализ CRISPR-локусов штаммов, не вошедших в исследованную выборку, а также сравнить полученные результаты с данными других методов типирования (например, MLST-типирования) возбудителя псевдотуберкулёза.
Заключение
Полученные результаты свидетельствуют о целесообразности использования CRISPR-типирования при мониторинге штаммов Y. pseudotuberculosis, циркулирующих на отдельных территориях, и выявления возможной связи структурно-функциональных характеристик сайтов CRISPR-Cas-систем с клиническими проявлениями псевдотуберкулёза. Имеющиеся данные определяют перспективу использования CRISPR-локусов в качестве специфических молекулярных маркеров при изучении внутривидового разнообразия и эволюции Y. pseudotuberculosis.
Источник финансирования. Авторы заявляют об отсутствии внешнего финансирования при проведении исследования.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
1 URL: https://omics.informatics.indiana.edu/CRISPRone/
2 URL: https://crisprcas.i2bc.paris-saclay.fr
3 URL: http://brabtools./CRISPRDetect
Об авторах
Надежда Павловна Перетолчина
Иркутский государственный медицинский университет
Автор, ответственный за переписку.
Email: nadine1lenz@gmail.com
ORCID iD: 0000-0001-9426-5197
SPIN-код: 2237-0140
Scopus Author ID: 57203299263
ResearcherId: P-8868-2017
м.н.с. НИИ биомедицинских технологий
Россия, ИркутскЕкатерина Александровна Воскресенская
Санкт-Петербургский научно-исследовательский институт эпидемиологии и микробиологии имени Пастера
Email: tsenevapasteur@yandex.ru
ORCID iD: 0000-0001-6380-1153
к.б.н., в.н.с. лаб. медицинской бактериологии
Россия, Санкт-ПетербургГалина Ивановна Кокорина
Санкт-Петербургский научно-исследовательский институт эпидемиологии и микробиологии имени Пастера
Email: tsenevapasteur@yandex.ru
ORCID iD: 0000-0002-1973-3138
к.м.н., с.н.с. лаб. медицинской бактериологии
Россия, Санкт-ПетербургЕлена Александровна Богумильчик
Санкт-Петербургский научно-исследовательский институт эпидемиологии и микробиологии имени Пастера
Email: tsenevapasteur@yandex.ru
ORCID iD: 0000-0002-3181-4328
н.с. лаб. медицинской бактериологии
Россия, Санкт-ПетербургВалерий Тимофеевич Климов
Иркутский научно-исследовательский противочумный институт Сибири и Дальнего Востока
Email: 41klimov@mail.ru
ORCID iD: 0000-0003-0036-0017
к.м.н., с.н.с. отдела эпидемиологии
Россия, ИркутскАлексей Леонидович Трухачев
Ростовский-на-Дону научно-исследовательский противочумный институт
Email: trukhachev_al@antiplague.ru
ORCID iD: 0000-0002-3531-1146
к.м.н., в.н.с. лаб. микробиологии чумы и других иерсиниозов
Россия, Ростов-на-ДонуЮрий Павлович Джиоев
Иркутский государственный медицинский университет
Email: alanir07@mail.ru
ORCID iD: 0000-0001-5410-5113
к.б.н., в.н.с. НИИ биомедицинских технологий
Россия, ИркутскВладимир Игоревич Злобин
Иркутский государственный медицинский университет; Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
Email: vizlobin@mail.ru
ORCID iD: 0000-0002-0164-5113
д.м.н., профессор, г.н.с. лаб. механизмов популяционной изменчивости патогенных микроорганизмов отдела арбовирусов и экспериментального производства Института вирусологии им. Д.И. Ивановского, профессор каф. микробиологии, вирусологии и иммунологии
Россия, Иркутск; МоскваСписок литературы
- Андрюков Б.Г., Сомова Л.М. У истоков экологической микробиологии: история открытия и изучения сапронозов. Здоровье. Медицинская экология. Наука. 2018;(1):55–62. Andryukov B.G., Somova L.M. Transformation of the sapronose representations at the new stage of integration of knowledge about them. Health, Medical Ecology, Science. 2018;(1):55–62. DOI: https://doi.org/10.5281/ZENODO.1194901 EDN: https://www.elibrary.ru/yssomm
- Bogdanovich T., Carniel E., Fukushima H., Skurnik M. Use of O-antigen gene cluster-specific PCRs for the identification and O-genotyping of Yersinia pseudotuberculosis and Yersinia pestis. J. Clin. Microbiol. 2003;41(11):5103–12. DOI: https://doi.org/10.1128/JCM.41.11.5103-5112.2003
- Климов В.Т., Чеснокова М.В. Молекулярно-генетический мониторинг Yersinia pseudotuberculosis на основе ПЦР-О-генотипирования. Молекулярная генетика, микробиология и вирусология. 2007;(4):14–7. Klimov V.T., Chesnokova M.V. Molecular genetic monitoring of Yersinia pseudotuberculosis using PCR O-genotyping. Molecular Genetics, Microbiology and Virology. 2007;(4):14–7. EDN: https://www.elibrary.ru/iccuij
- Кокорина Г.И., Сайнес Т.В., Богумильчик Е.А., Воскресенская Е.А. Особенности эпидемического процесса и лабораторной диагностики иерсиниозов в Российской Федерации в 2016–2018 гг. Проблемы медицинской микологии. 2022;24(2):86. Kokorina G.I., Saines T.V., Bogumil'chik E.A., Voskresenskaya E.A. Features of the epidemic process and laboratory diagnosis of Yersiniosеs in the Russian Federation in 2016-2018. Problems in Medical Mycology. 2022;24(2):86. EDN: https://www.elibrary.ru/qbgywj
- Чеснокова М.В., Климов В.Т., Никитин А.Я. и др. Анализ эпидемиологической ситуации по псевдотуберкулезу и кишечному иерсиниозу в России и прогноз заболеваемости на среднесрочную перспективу. Здоровье населения и среда обитания – ЗНиСО. 2018;(9):59–64. Chesnokova M.V., Klimov V.T., Nikitin A.Ya., et al. Analysis of the epidemiological situation on pseudotuberculosis and intestinal yersiniosis in Russia and forecast of the incidence rate in the medium-term perspective. Public Health and Life Environment – PH&LE. 2018;(9):59–64. DOI: https://doi.org/10.35627/2219-5238/2018-306-9-59-64 EDN: https://www.elibrary.ru/vagnaj
- Laukkanen-Ninios R., Didelot X., Jolley K.A., et al. Population structure of the Yersinia pseudotuberculosis complex according to multilocus sequence typing: MLST of the Y. pseudotuberculosis complex. Environ. Microbiol. 2011; 13(12): 3114–27. DOI: https://doi.org/10.1111/j.1462-2920.2011.02588.x
- Мелоян М.Г., Воскресенская Е.А., Лебедева С.А., Трухачев А.Л. Обзор методов генетической дифференциации штаммов возбудителя псевдотуберкулеза. Вестник Пермского университета. Серия: Биология. 2022;(1):54–63. Meloyan M.G., Voskresenskaya E.A., Lebedeva S.A., Trukhachev A.L. Review of the methods of genetic differentiation of pseudotuberculosis pathogen strains. Bulletin of Perm University. Series Biology. 2022;(1):54–63. DOI: https://doi.org/10.17072/1994-9952-2022-1-54-63 EDN: https://www.elibrary.ru/roxmqp
- Grissa I., Vergnaud G., Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. DOI: https://doi.org/10.1186/1471-2105-8-172
- Koskela K.A., Mattinen L., Kalin-Mänttäri L., et al. Generation of a CRISPR database for Yersinia pseudotuberculosis complex and role of CRISPR-based immunity in conjugation: Y. pseudotuberculosis complex CRISPR database. Environ. Microbiol. 2015;17(11):4306–21. DOI: https://doi.org/10.1111/1462-2920.12816
- Seecharran T., Kalin-Manttari L., Koskela K., et al. Phylogeographic separation and formation of sexually discrete lineages in a global population of Yersinia pseudotuberculosis. Microb. Genom. 2017;3(10):e000133. DOI: https://doi.org/10.1099/mgen.0.000133
- Перетолчина Н.П., Климов В.Т., Воскресенская Е.А. и др. Локусный состав CRISPR-Cas системы Yersinia pseudotuberculosis различных генетических вариантов. Эпидемиология и вакцинопрофилактика. 2020;19(2):31–9. Peretolchina N.P., Klimov V.T., Voskresenskaya E.A., et al. CRISPR-Cas loci of Yersinia pseudotuberculosis strains with different genetic determinants. Epidemiology & Vaccinal Prevention. 2020;19(2):31–9. DOI: https://doi.org/10.31631/2073-3046-2020-19-2-31-39 EDN: https://www.elibrary.ru/zqepfb
- Кокорина Г.И. Генотипы штаммов Yersinia pseudotuberculosis и их клинические и диагностичеcкое значение: Автореф. дисс. … канд. мед. наук. СПб.; 2012. Kokorina G.I. Genotypes of Yersinia pseudotuberculosis strains and their clinical and diagnostic significance: Diss. St. Petersburg; 2012.
- Galardini M., Biondi E.G., Bazzicalupo M., Mengoni A. CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol. Med. 2011;6:11. https://doi.org/10.1186/1751-0473-6-11
- Prjibelski A., Antipov D., Meleshko D., et al. Using SPAdes de novo assembler. Curr. Protoc. Bioinformatics. 2020;70(1):e102. https://doi.org/10.1002/cpbi.102
- Biswas A., Staals R.H.J., Morales S.E., et al. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17:356. DOI: https://doi.org/10.1186/s12864-016-2627-0
- Zhang Q., Ye Y. Not all predicted CRISPR–Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics. 2017;18(1):92. DOI: https://doi.org/10.1186/s12859-017-1512-4
- Couvin D., Bernheim A., Toffano-Nioche C., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46(W1):W246–51. DOI: https://doi.org/10.1093/nar/gky425
- McGinnis S., Madden T.L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32(Issue suppl. 2):W20–5. DOI: https://doi.org/10.1093/nar/gkh435
- Biswas A., Gagnon J.N., Brouns S.J.J., et al. CRISPRTarget: Bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10(5):817–27. DOI: https://doi.org/10.4161/rna.24046
- Arndt D., Grant J.R., Marcu A., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44(W1):W16–21. DOI: https://doi.org/10.1093/nar/gkw387
- R Core Team. R: A Language and Environment for Statistical Com- puting. Vienna: R Foundation for Statistical Computing; 2021.
- Oksanen J., Simpson G., Blanchet G., et al. Vegan: Community Ecology Package; 2022. URL: https://cran.r-project.org/web/packages/vegan/vegan.pdf
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–9. DOI: https://doi.org/10.1093/bioinformatics/btw313
- Zhou Y., Liang Y., Lynch K.H., et al. PHAST: a fast phage search tool. Nucleic Acids Res. 2011; 39(Suppl.): W347–52. DOI: https://doi.org/10.1093/nar/gkr485
- Westra E.R., Buckling A., Fineran P.C. CRISPR–Cas systems: beyond adaptive immunity. Nat. Rev. Microbiol. 2014;12(5):317–26. DOI: https://doi.org/10.1038/nrmicro3241
- Medina-Aparicio L., Dávila S., Rebollar-Flores J.E., et al. The CRISPR-Cas system in Enterobacteriaceae. Pathog. Dis. 2018;76(1). DOI: https://doi.org/10.1093/femspd/fty002
- Horvath P., Romero D.A., Coûté-Monvoisin A.C., et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 2008;190(4):1401–12. DOI: https://doi.org/10.1128/JB.01415-07
- Vergnaud G., Zhou D., Platonov M.E., et al. Analysis of the three Yersinia pestis CRISPR loci provides new tools for phylogenetic studies and possibly for the investigation of ancient DNA. In: Perry R.D., Fetherston J.D., eds. The Genus Yersinia. New York: Springer New York; 2007:327–38. DOI: https://doi.org/10.1007/978-0-387-72124-8_30
- Mokrousov I., Limeschenko E., Vyazovaya A., Narvskaya O. Corynebacterium diphtheriae spoligotyping based on combined use of two CRISPR loci. Biotechnol. J. 2007;2(7):901–6. DOI: https://doi.org/10.1002/biot.200700035
- Yosef I., Goren M.G., Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40(12):5569–76. DOI: https://doi.org/10.1093/nar/gks216
- Garrett S.C. Pruning and tending immune memories: spacer in the CRISPR array. Front. Microbiol. 2021;12:664299. DOI: https://doi.org/10.3389/fmicb.2021.664299
- Bertelli C., Tilley K.E., Brinkman F.S.L. Microbial genomic island discovery, visualization and analysis. Brief. Bioinform. 2019;20(5):1685–98. DOI: https://doi.org/10.1093/bib/bby042
- Wimmer F., Beisel C.L. CRISPR-Cas systems and the paradox of self-targeting spacers. Front. Microbiol. 2020;10:3078. DOI: https://doi.org/10.3389/fmicb.2019.03078.
Дополнительные файлы

