


Оптимизация мультилокусного сиквенс-анализа для лабораторной идентификации возбудителей иксодового клещевого боррелиоза
- Авторы: Голидонова К.А.1, Коренберг Э.И.1, Гинцбург А.Л.1
-
Учреждения:
- Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
- Выпуск: Том 99, № 5 (2022)
- Страницы: 514-524
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://microbiol.crie.ru/jour/article/view/1339
- DOI: https://doi.org/10.36233/0372-9311-296
- ID: 1339

Цитировать

Аннотация
Введение. Наиболее широко распространённые этиологические агенты иксодового клещевого боррелиоза (ИКБ) в России — Borrelia garinii, B. afzelii, B. bavariensis. Для определения видовой принадлежности боррелий в современных исследованиях используют методы мультилокусного сиквенс-типирования и сиквенс-анализа (МЛСА). Ранее были продемонстрированы результаты применения схемы МЛСА для идентификации возбудителей эритемных форм ИКБ.
Цель работы — изучить возможность оптимизации МЛСА для практической лабораторной идентификации возбудителей ИКБ. Задачи: сравнительный анализ нуклеотидных последовательностей локусов 6 консервативных генов (rrs, hbb, fla, groEL, recA, ospA) и межгенного спейсера rrfA-rrlB, рекомендованных протоколом МЛСА; выявление минимальной совокупности генов, сцепленные сиквенсы которых позволяют идентифицировать видовую принадлежность изолята боррелий.
Материалы и методы. Сиквенсы вышепредставленных локусов 23 «контрольных» изолятов, полученных от больных ИКБ и предварительно типированных методом МЛСА как B. bavariensis, использованы для сравнительного анализа с последовательностями аналогичных генов других видов боррелий, имеющимися в международных базах данных. На основе этого материала методом UPGMA построена и проанализирована серия дендрограмм.
Результаты. Сиквенсы локусов гена ospA контрольного вида показали наибольшее отличие (не менее 8,5%) от последовательностей этого гена у других сравниваемых видов боррелий; близкие показатели видовых отличий (не менее 6,7%) продемонстрировало сравнение сиквенсов гена recA. Сиквенсы выявленных вариантов двух этих генов у B. bavariensis отличались от последовательностей аналогичных генов у наиболее близкого вида — B. garinii. Дендрограмма сцепленных нуклеотидных последовательностей генов recA и ospA продемонстрировала её идентичность результатам идентификации изолятов по полному протоколу МЛСА.
Заключение. Предложен оптимизированный подход к МЛСА боррелий группы B. burgdorferi sensu lato, который сводится к выявлению их видовой принадлежности на основании специфики результата сцепленного анализа локусов только двух генов (recA и ospA) из 7 локусов, рекомендованных протоколом этого метода.
Ключевые слова
Полный текст
Введение
Иксодовые клещевые боррелиозы (ИКБ) — группа этиологически самостоятельных хронических или рецидивирующих спирохетозных природно-очаговых трансмиссивных инфекций, способных поражать различные системы и органы. Природные очаги ИКБ распространены в России в основном в лесной зоне. По уровню заболеваемости боррелиозы занимают одно из ведущих мест среди природно-очаговых зоонозов [1, 2].
Возбудители ИКБ — спирохеты комплекса Borrelia burgdorferi sensu lato, включающего более 20 видов [3], 8 из которых обнаружены в России [4]. Наиболее широко распространённые этиологические агенты ИКБ в нашей стране и большей части Евразии — B. garinii, B. afzelii и B. bavariensis [1, 5]. B. garinii чаще вызывает среднетяжёлую форму заболевания с общим инфекционным синдромом, при этом около 60% случаев составляет эритемная форма. Инфекционный процесс, вызванный B. afzelii, чаще протекает в лёгкой форме и обычно сопровождается развитием мигрирующей эритемы на месте укуса клеща [6, 7]. Эритемную форму ИКБ с различным спектром клинических проявлений вызывает и B. bavariensis [5]. Следовательно, мигрирующая эритема, которую клиницисты считают единственным патогномоничным признаком ИКБ, с большей или меньшей частотой может возникать при любом из наиболее распространённых этиологических агентов заболеваний этой группы.
Даже при наличии у пациента эритемы, степень выраженности и величина которой могут зависеть от ряда причин, включая сходные кожные проявления иной этиологии, клинический диагноз ИКБ в большинстве случаев нуждается в лабораторном подтверждении. С этой целью в настоящее время обычно применяют различные серологические методы: реакцию непрямой иммунофлюоресценции, иммуноферментный анализ, иммуноблоттинг, варианты мультиплексного анализа (включая фосфоресцентный) и др. Эти методы характеризуются различными показателями специфичности и чувствительности, варьирующими в разные сроки от начала заболевания ИКБ. Они, как правило, наименее эффективны в его первые недели и совершенно безрезультативны в нередких серонегативных случаях. Возможность прямой изоляции возбудителя путём посевов проб на питательную среду сильно ограничена непродолжительностью боррелиемии, а также необязательностью его присутствия (особенно в остром периоде инфекционного процесса) в том или ином органе, тканях и биологических жидкостях пациента. Кроме того, посевы биоматериала не могут быть массовым лабораторно-диагностическим тестом ИКБ из-за продолжительности роста боррелий в положительных пробах. Однако из различных материалов, полученных от пациентов, можно амплифицировать ДНК боррелий методом полимеразной цепной реакции (ПЦР). Её клиническая чувствительность при исследовании ликвора пациентов не превышает 20%, при анализе плазмы крови — варьирует от 30 до 50%, при тестировании синовиальной жидкости может превышать 70%, а биоптатов кожи — 80% [1, 8].
Для определения видовой принадлежности боррелий в современных исследованиях широко используют методы мультилокусного сиквенс-типирования (МЛСТ) [9] и сиквенс-анализа (МЛСА) [1], основанные на выявлении специфики нуклеотидных последовательностей консервативных генов [10]. Применительно к видовой идентификации этиологии ИКБ каждый из этих методов имеет определённые особенности. Так, оба метода основаны на анализе нуклеотидных последовательностей локусов 7–8 разных консервативных генов (включая межгенный спейсер rrfA-rrlB), которые рекомендовано использовать при построении итоговых дендрограмм по сцепленным данным. При МЛСТ для построения дендрограмм используют матрицу несовпадений в аллельном профиле, игнорируя количество нуклеотидных различий между аллелями, которым присваивают разные номера независимо от того, различаются ли нуклеотидные последовательности в одном сайте или во многих сайтах [10]. Этот метод давно и успешно используют при видовой идентификации различных групп патогенных бактерий, а для некоторых из них его считают «золотым стандартом» [11–13]. При МЛСА, наоборот, для определения филогенетических отношений тестируемых образцов используют сцепленные последовательности фрагментов их генов домашнего хозяйства [4, 10]. Схемой МЛСТ в зависимости от источника и концентрации ДНК рекомендовано проводить одно- или двухраундовую ПЦР и амплифицировать более длинные фрагменты консервативных генов [14], а схема МЛСА рекомендует однораундовую ПЦР менее длинных локусов [10, 15]. Поэтому для видовой идентификации значительного числа изолятов боррелий МЛСА во всех отношениях менее затратен, чем МЛСТ. Подробнее преимущества и недостатки этих методов рассмотрены нами ранее [16].
Результаты применения схемы МЛСА для идентификации этиологического агента эритемных форм ИКБ продемонстрированы нами на клиническом материале (более 20 изолятов, полученных из биоптатов кожи и плазмы крови пациентов). Было установлено, что все изоляты относятся к B. bavariensis, выявлена вариабельность нуклеотидных последовательностей локусов анализированных консервативных генов [5]. Это определило цель нашего исследования: изучить возможность оптимизации МЛСА для практической лабораторной идентификации возбудителей ИКБ.
Поставлены следующие задачи: сравнительный количественный анализ внутривидовой гетерогенности нуклеотидных последовательностей локусов 6 консервативных хромосомных генов (rrs, hbb, fla, groEL, recA, ospA) и межгенного спейсера (rrfA-rrlB), рекомендованных для МЛСА; выявление минимальной совокупности генов, сцепленные нуклеотидные последовательности которых позволяют идентифицировать видовую принадлежность изолята боррелий группы B. burgdorferi sensu lato.
Материалы и методы
У 23 изолятов, хранящихся в музейной коллекции боррелий лаборатории переносчиков инфекций ФГБУ НИЦЭМ имени почётного академика Н.Ф. Гамалеи, исследованы нуклеотидные последовательности локусов каждого из 6 генов (перечислены выше) и спейсера rrfA-rrlB, рекомендованных для МЛСА [15]. Изоляты получены от больных людей путём посева на среду BSK кожных биоптатов из периферической части эритем (17 изолятов) или из плазмы крови (6 изолятов) пациентов Краевой инфекционной больницы г. Пермь (восток Восточной Европы; 58°00' с.ш.; 56°15' в.д.) с локализованной стадией манифестной формы ИКБ с мигрующей эритемой. Количество исследованных изолятов составляет не менее 30–40% среднего числа всех пациентов с ИКБ, ежегодно поступающих в этот клинический стационар [6]. Культивирование и типирование изолятов методом ПЦР-полиморфизма длин рестрикционных фрагментов спейсерного участка rrfA-rrlB позволило ранее отнести их к B. garinii NТ29 [17, 18]. По результатам наших исследований методом МЛСА эти изоляты идентифицированы как B. bavariensis. Нуклеотидные последовательности использованных праймеров, режим ПЦР и условия последующего секвенирования ампликонов описаны ранее [5, 15, 19].
Все полученные нуклеотидные последовательности сравнивали между собой (с использованием платформы BLAST) и с нуклеотидными последовательностями локусов аналогичных генов у типовых или референсных штаммов других видов спирохет B. burgdorferi sensu lato, представленными в базах данных GenBank INSDC и PubMLST Borrelia spp. Для этого методом невзвешенного попарного среднего (UPGMA) была построена серия дендрограмм с использованием пакета программ «MEGA-X v. 10.2.4» при величине bootstrap 1000 повторов. Значительная часть этих дендрограмм проанализирована ранее [5, 19]. В данной статье приведены лишь те, которые необходимы для обоснования заключения данной статьи и ранее не публиковались.
Нуклеотидные последовательности 10 локусов исследованных генов депонированы в базу данных Genbank (номера доступа MW981426, MZ005315-MZ005321, OM310938–OM310939) и 4 более коротких сиквенса — в базу данных European Nucleotide Archive (номера доступа OD916881–OD916884).
Результаты
Сравнительный анализ данных нуклеотидных последовательностей локусов генов, рекомендованных для МЛСА
В табл. 1 представлены показатели сходства нуклеотидных последовательностей локусов каждого гена у всех изученных нами изолятов, идентифицированных по результатам МЛСА как B. bavariensis, включая опубликованные ранее данные по генам rrs, fla, hbb и recA [5, 19]. Сходство оказалось значительным (до 99,5–100%) у изолятов из кожных биоптатов и из плазмы крови пациентов. С локусами других генов минимальное сходство (95,3%) имели некоторые последовательности гена ospA. Однако сиквенсы этого гена на 9,5–10,9% отличались от сиквенсов того же гена B. garinii 20047T (наиболее близкий вид по общей структуре генома [20]), а также на 8,5% (т.е. больше, чем у всех других генов) от совокупности сиквенсов широко распространённых боррелий группы B. burgdorferi sensu lato. Аналогичные показатели для сиквенсов гена recA оказались несколько меньше, чем у гена ospA, но больше, чем у остальных исследованных генов (табл. 1). Отличия нуклеотидных последовательностей секвенированных нами локусов двух этих генов от последовательности аналогичного участка генома референсного штамма PBi европейской подгруппы B. bavariensis были существенными (до 8,1%). Эти результаты стимулировали проведение более детального анализа внутривидовой гетерогенности нуклеотидных последовательностей генов recA и ospA на материале наших изолятов B. bavariensis, а также их отличий от сиквенсов аналогичных генов у боррелий других видов комплекса B. burgdorferi sensu lato.
Таблица 1. Вариабельность нуклеотидных последовательностей совокупности локусов (по результатам 23 исследованных изолятов B. bavariensis) каждого гена
Table 1. Variability of the nucleotide sequences of the set of loci (according to the results of 23 studied isolates of B. bavariensis) of each gene
Ген (мишень) Gene (target) | Сходство нуклеотидных последовательностей между изолятами из кожных биоптатов и из плазмы, % Nucleotide sequence similarity between skin biopsy and plasma isolates, % | Отличие от (в %) | Difference from (in %) | ||
B. bavariensis PBi | B. garinii 20047T | других видов боррелий B. burgdorferi s. l, не менее other Borrelia species B. burgdorferi s. l, not less than | ||
rrs | 100 | 0,4 | 0,2 | 0,8 |
hbb | 98,5–100 | 1,2–1,5 | 1,2–1,5 | 4,3 |
fla | 99,5–100 | 0,6–1,2 | 1,2–1,8 | 5,8 |
groEL | 98,7–100 | 2,2–2,7 | 2,2–2,7 | 3,6 |
recA | 97,3–100 | 0,7–2 | 2–3,4 | 6,7 |
ospA | 95,3–100 | 4,7–8,1 | 9,5–10,9 | 8,5 |
Спейсер rrfA-rrlB Spacer rrfA-rrlB | 98,8–100 | 1,7–2,3 | 1,7–2,3 | 4,8 |
Сцепленные последовательности локусов генов recA и ospA Concatenated sequences of recA and ospA gene loci | 96,1–100 | 3,6–5,6 | 6,4–7,8 | 8,3 |
Детальный анализ дендрограмм последовательностей локусов генов recA и ospA и сравнение этих данных с результатами полного МЛСА
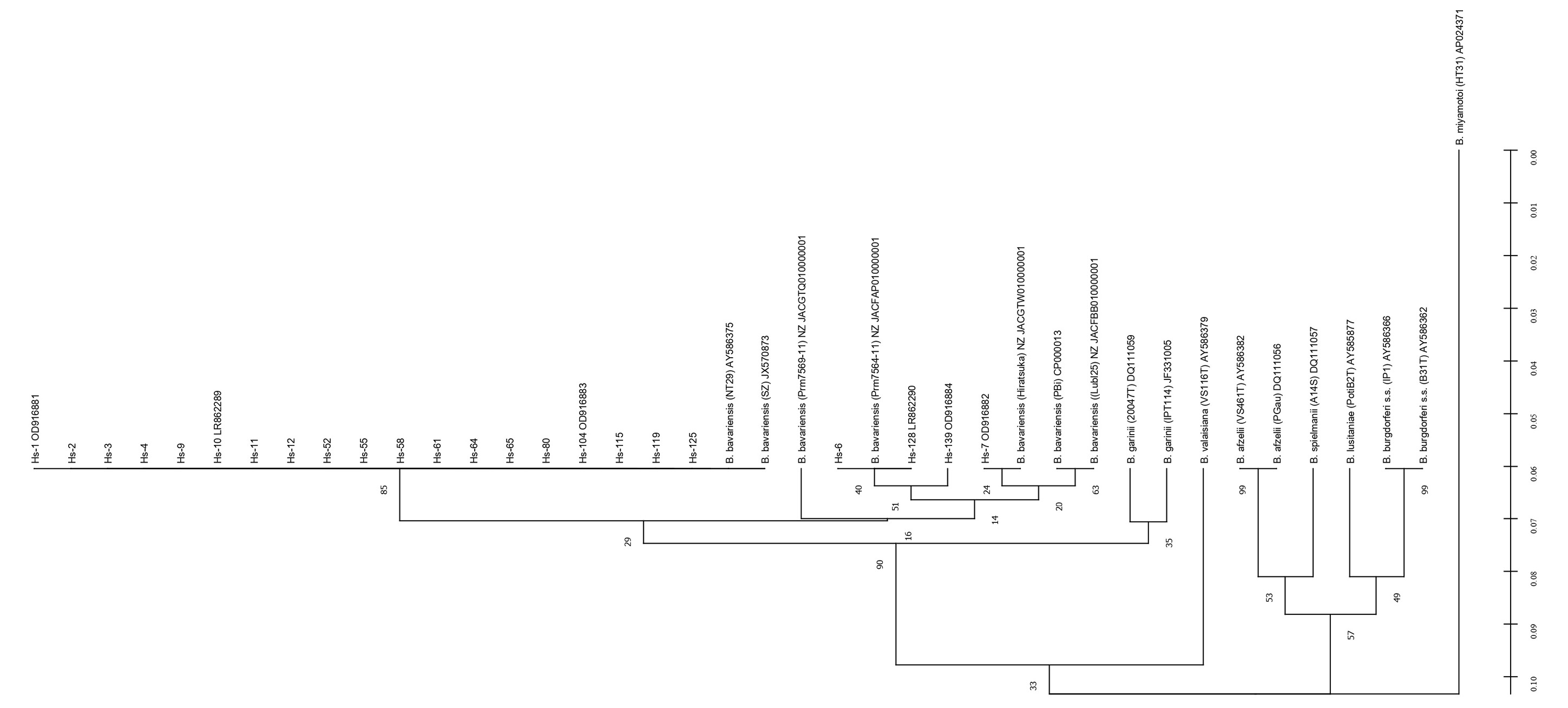
Сиквенсы генов recA анализированных нами изолятов показали, что все они попадают в большой кластер с тремя различными аллельными вариантами евразийской генетической подгруппы B. bavariensis: нуклеотидные последовательности большинства изолятов (19 из 23) идентичны варианту SZ, 3 — варианту Prm7504-11 и 1 — варианту Hiratsuka (номера доступа GenBank; рис. 1). Это объясняет некоторое несходство нуклеотидных последователей локусов этого гена у разных изолятов (табл. 1). Тем не менее все сиквенсы этого гена чётко отличаются от такового у B. garinii и референсных штаммов наиболее распространённых видов B. burgdorferi sensu lato, а также от последовательности этого гена у широко распространённой патогенной B. miyamotoi, таксономический статус которой остаётся дискуссионным [1]. Аналогичная дендрограмма, построенная по результатам секвенирования локусов гена ospA (рис. 2), в целом принципиально не отличается от предыдущей.

Рис. 1. Дендрограмма нуклеотидных последовательностей гена recA исследованных изолятов. Здесь и на рис. 2, 3 в круглых скобках приведено наименование штаммов по базе данных PubMLST Borrelia spp.; в квадратных скобках — номера доступа в банках Genbank или ENA. Hs — изоляты от пациентов. Величины bootstrap (1000) указаны в процентах около соответствующего узла. Для определения коэффициента конгруэнтности между матрицами генетических расстояний генов recA и ospA по отдельности и в сопоставлении с матрицей сцепленных последовательностей применён тест Мантеля [24] в «Excel» с надстройкой GenALEx — R = 0,499 (recA/ospA).
Здесь и на рис. 2, 3 в круглых скобках приведено наименование штаммов по базе данных PubMLST Borrelia spp.; в квадратных скобках — номера доступа в банках Genbank или ENA. Hs — изоляты от пациентов. Величины bootstrap (1000) указаны в процентах около соответствующего узла. Для определения коэффициента конгруэнтности между матрицами генетических расстояний генов recA и ospA по отдельности и в сопоставлении с матрицей сцепленных последовательностей применён тест Мантеля [24] в «Excel» с надстройкой GenALEx — R = 0,499 (recA/ospA).

Рис. 2. Дендрограмма нуклеотидных последовательностей гена ospA. Тест Мантеля — R = 0,838 (ospA/recA).
Тест Мантеля — R = 0,838 (ospA/recA).
Результаты МЛСА 6 отобранных изолятов, которые представляют все выявленные нами аллельные варианты генов recA и ospA B. bavariensis (рис. 1, 2), показали, что они на 98,5–100,0% сходны с сиквенсами различных изолятов евразийской генетической подгруппы боррелий этого вида (SZ, Prm7564-11, Prm7569-11, Hiratsuka) и несколько менее — с европейскими изолятами, например, с референсным штаммом PBi — на 97,8–98,4%. Вместе с тем все генетические варианты B. bavariensis, включая штамм PBi, имеют существенные отличия (т.е. сходство не более 94,9%), позволяющие четко дифференцировать их по данным МЛСА от боррелий других видов (табл. 2). Дендрограмма сцепленных нуклеотидных последовательностей двух генов (recA и ospA) тех же наших изолятов демонстрирует наличие двух генетических подгрупп B. bavariensis (евразийской и европейской) и их чёткие отличия от соответствующих сцепленных сиквенсов боррелий других видов из баз данных (рис. 3).
Таблица 2. Сходство (в %) некоторых «контрольных» изолятов с боррелиями различных видов, нуклеотидные последовательности которых имеются в базах данных GenBank и PubMLST
Table 2. Similarity (in %) of some “control” isolates with Borrelia of various species, the nucleotide sequences of which are available in the GenBank and PubMLST databases
Hs-7 | Hs-10 | Hs-55 | Hs-104 | Hs-128 | Hs-139 | B. garinii 20047T | B. bavariensis | B. spielmanii A14S | B. burgdorferi s. s. B31T | B. lusitaniae PotiB2T | B. valaisiana VS116T | B. afzelii VS461T | |||||
PBi | NT 29 | Prm7564-11 | Hiratsuka | ||||||||||||||
Hs-7 | 100 | ||||||||||||||||
Hs-10 | 98,9 | 100 | |||||||||||||||
Hs-55 | 98,9 | 100 | 100 | ||||||||||||||
Hs-104 | 98,9 | 100 | 100 | 100 | |||||||||||||
Hs-128 | 100 | 98,9 | 98,8 | 98,9 | 100 | ||||||||||||
Hs-139 | 99,6 | 98,9 | 98,8 | 98,9 | 99,7 | 100 | |||||||||||
B. garinii 20047T | 97,8 | 97,6 | 97,5 | 97,6 | 97,7 | 97,3 | 100 | ||||||||||
B. bavariensis | PBi | 98,2 | 98,4 | 98,3 | 98,4 | 98,1 | 97,8 | 98,1 | 100 | ||||||||
NT29 | 98,8 | 100 | 99,9 | 100 | 98,8 | 98,8 | 97,6 | 98,4 | 100 | ||||||||
Prm7564-11 | 100 | 98,9 | 98,8 | 98,9 | 100 | 99,7 | 97,7 | 98,1 | 98,8 | 100 | |||||||
Hiratsuka | 100 | 98,9 | 98,9 | 98,9 | 100 | 99,6 | 97,8 | 98,2 | 98,8 | 100 | 100 | ||||||
B. spielmanii A14S | 94,5 | 94,5 | 94,5 | 94,5 | 94,4 | 94,4 | 94,3 | 94,0 | 94,5 | 94,4 | 94,5 | 100 | |||||
B. burgdorferi s. s. B31T | 93,6 | 93,5 | 93,4 | 93,5 | 93,5 | 93,5 | 93,3 | 93,2 | 93,5 | 93,5 | 93,6 | 93,6 | 100 | ||||
B. lusitaniae PotiB2T | 93,8 | 93,9 | 93,9 | 93,9 | 93,7 | 93,9 | 93,6 | 93,5 | 93,9 | 93,7 | 93,8 | 94 | 93,3 | 100 | |||
B. valaisiana VS116T | 94,5 | 94,9 | 94,9 | 94,9 | 94,5 | 94,4 | 94,2 | 94,4 | 94,6 | 94,5 | 94,5 | 94,3 | 93,8 | 94,3 | 100 | ||
B. afzelii VS461T | 93,9 | 93,9 | 93,8 | 93,9 | 93,9 | 94,0 | 94,2 | 94,0 | 93,9 | 93,9 | 93,9 | 94,5 | 94,1 | 93,1 | 94,2 | 100 | |

Рис. 3. Дендрограмма сцепленных нуклеотидных последовательностей генов recA и ospA. Тест Мантеля — R = 0,860 (сцепленные/recA), R = 0,965 (сцепленные/ospA).
Тест Мантеля — R = 0,860 (сцепленные/recA), R = 0,965 (сцепленные/ospA).
Это свидетельствует о полной идентичности результатов идентификации боррелий группы B. bur- gdorferi sensu lato путём анализа сцепленных нуклеотидных последовательностей локусов генов recA, ospA и по протоколу МЛСА.
Обсуждение
Определение видовой принадлежности боррелий группы B. burgdorferi sensu lato, циркулирующих в природных очагах и (или) вызвавших заболевание ИКБ, как было отмечено выше, в настоящее время в основном проводится молекулярно-биологическими методами. При этом использование в качестве мишени какого-либо одного консервативного гена или спейсера обычно не позволяет точно идентифицировать изучаемый образец [10, 20]. Методы МЛСА и МЛСТ, протоколами которых предусмотрено секвенирование локусов нескольких (6–8) генов, затратны, времяёмки и затруднительны [5, 10, 14–16], особенно при необходимости быстрой индикации этиологического агента ИКБ в условиях диагностических лабораторий лечебных учреждений.
Представленный в этой статье детальный анализ данных, полученных ранее и характеризующих генетическую структуру пула изолятов B. bavariensis [5, 19], привёл к выводу, что нуклеотидные последовательности локусов генов recA и ospA в наибольшей степени отличаются от последовательностей других генов, рекомендованных протоколом МЛСА для индикации распространённых видов комплекса B. burgdorferi sensu lato. Ранее были предприняты попытки достижения этой цели на основе разнообразия структуры отдельно каждого из генов recA и ospA [21, 22]. Сравнение таких наших данных (рис. 1 и 2) показывает, что они дают близкие, но не идентичные результаты генотипической принадлежности некоторых изолятов. Однако дендрограмма сцепленных нуклеотидных последовательности локусов генов recA и ospA (рис. 3) демонстрирует точно такую же видовую и геновариантную принадлежность изолятов, как и ранее полученные результаты их типирования в соответствии с полным протоколом МЛСА.
Для проведения «контрольного» исследования, соответствующего задаче статьи, нами использован репрезентативный пул изолятов B. bavariensis, видовая принадлежность и гетерогенность которых были предварительно изучены этим методом и известны [5]. Сцепленные нуклеотидные последовательности локусов генов recA и ospA, принадлежащие референсным штаммам каждого из наиболее распространённых видов группы B. burgdorferi sensu lato (рис. 3), чётко различаются. Поэтому видовая принадлежность вновь исследуемого образца может быть выявлена путём расчёта максимального сходства сцепленных последовательностей этих двух его генов с аналогичными последовательностями определённых видов боррелий данной группы, включая B. bavariensis, или построением аналогичной дендрограммы. Для оптимизации регулярных лабораторных исследований её неизменный «шаблон» по типу рис. 3 может храниться в виде файла (например, в программе MEGA или другой). Простое сопоставление числа генов, рекомендованных протоколами МЛСА и МЛСТ, с их числом, которое по представленным нами данным необходимо и достаточно секвенировать для определения уже известного науке вида возбудителя ИКБ, показывает, что трудовые и финансовые затраты могут быть при этом сокращены примерно в 3–4 раза.
Специального дальнейшего изучения заслуживает возможность дифференциации боррелий группы B. burgdorferi sensu lato от B. miyamotoi по их нуклеотидным последовательностям гена recA, существенные отличия которых подтверждают наши данные (рис. 1). Это может иметь важное значение для совершенствования генодиагностики этиологии ИКБ, тем более что известен прецедент использования этого гена в совокупности с двумя другими для лабораторного подтверждения заболевания, вызванного B. miyamotoi [23].
Заключение
На основании исследований, приведённых выше, мы предлагаем оптимизированный подход к МЛСА боррелий группы B. burgdorferi sensu lato. Он сводится к выявлению их видовой принадлежности на основании специфики результата сцепленного анализа локусов только 2 генов (recA и ospA) из 6, а также спейсера rrfA-rrlB, рекомендованных протоколом данного метода. Это значительно сокращает затраты и ускоряет лабораторное исследование идентифицируемых образцов.
Об авторах
Кристина Андреевна Голидонова
Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
Автор, ответственный за переписку.
Email: kristi.dekor@mail.ru
ORCID iD: 0000-0003-4832-6248
н.с.
Россия, МоскваЭдуард Исаевич Коренберг
Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
Email: kristi.dekor@mail.ru
ORCID iD: 0000-0002-4452-4231
д.б.н.; профессор, зав. лаб. переносчиков инфекций
Россия, МоскваАлександр Леонидович Гинцбург
Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
Email: kristi.dekor@mail.ru
ORCID iD: 0000-0003-1769-5059
д.б.н.; профессор, академик РАН; директор ФГБУ НИЦЭМ им. почётного академика Н.Ф. Гамалеи
Россия, МоскваСписок литературы
- Коренберг Э.И., Помелова В.Г., Осин Н.С. Природноочаговые инфекции, передающиеся иксодовыми клещами. М.: Комментарий; 2013.
- Рудакова С.А., Пеньевская Н.А., Блох А.И., Рудаков Н.В., Транквилевский Д.В., Савельев Д.А. и др. Обзор эпидемиологической ситуации по иксодовым клещевым боррелиозам в Российской Федерации в 2010–2020 гг. и прогноз на 2021 г. Проблемы особо опасных инфекций. 2021; (2): 52–61. https://doi.org/10.21055/0370-1069-2021-2-52-61
- Margos G., Castillo-Ramirez S., Cutler S., Dessau R.B., Eikeland R., Estrada-Peña A., et al. Rejection of the name Borreliella and all proposed species comb. nov. placed therein. Int. J. Syst. Evol. Microbiol. 2020; 70(5): 3577–81. https://doi.org/10.1099/ijsem.0.004149
- Нефедова В.В., Коренберг Э.И., Горелова Н.Б. Мультилокусный сиквенс-анализ "нетипичных" Borrelia burgdorferi sensu lato, изолированных в России. Молекулярная генетика, микробиология и вирусология. 2017; 35(4): 145–50. https://doi.org/10.3103/S0891416817040073
- Голидонова К.А., Коренберг Э.И., Горелова Н.Б., Гинцбург А.Л. Мультилокусный сиквенс-анализ изолятов от больных эритемной формой иксодового клещевого боррелиоза. Молекулярная генетика, микробиология и вирусология. 2021; 39(4): 14–20. https://doi.org/10.3103/S089141682104008X
- Тетерин В.Ю., Коренберг Э.И., Нефедова В.В., Воробьева Н.Н., Фризен В.И., Помелова В.Г. и др. Клинико-лабораторная диагностика инфекций, передающихся иксодовыми клещами, в Пермском крае. Эпидемиология и инфекционные болезни. 2013; (4): 11–5.
- Фоменко Н.В., Романова Е.В., Караваева Ю.Ю., Панов В.В., Черноусова Н.Я., Ливанова Н.Н. Разнообразие Borrelia burgdorferi sensu lato в природных очагах Новосибирской области. Бюллетень Сибирской медицины. 2006; 5(S1): 93–8. https://doi.org/10.20538/1682-0363-2006-5-S1-93-98
- Branda J.A., Steere A.C. Laboratory diagnosis of Lyme borreliosis. Clin. Microbiol. Rev. 2021; 34(2): e00018-19. https://doi.org/10.1128/CMR.00018-19
- Платонов А.Е., Шипулин Г.А., Платонова О.В. Мультилокусное секвенирование – новый метод генотипирования бактерий и первые результаты его применения. Генетика. 2000; 36(5): 597–605.
- Radolf J., Samuels S. Lyme Disease and Relapsing Fever Spirochetes: Genomics, Molecular Biology, Host Interactions and Disease Pathogenesis. Caister Academic Press Limited; 2021. https://doi.org/10.21775/9781913652616
- Платонов А.Е., Миронов К.О., Яцышина С.Б., Королева И.С., Платонова О.В., Гущин А.Е. и др. Характеристика московских штаммов Haemophilus influenzae типа b методом мультилокусного секвенирования-типирования. Молекулярная генетика, микробиология и вирусология. 2003; (2): 21–5.
- Мухачева Т.А., Ковалев С.Ю. Мультилокусное сиквенс-типирование боррелий на территории России: промежуточные итоги и перспективы. Национальные приоритеты России. 2014; (3): 110–3.
- Слукин П.В., Асташкин Е.И., Асланян Е.М., Ершова М.Г., Полетаева Е.Д., Светоч Э.А. и др. Характеристика вирулентных штаммов Escherichia coli, выделенных от пациентов с урологической инфекцией. Журнал микробиологии, эпидемиологии и иммунобиологии. 2021; 98(6): 671–84. https://doi.org/10.36233/0372-9311-134
- Margos G., Gatewood A.G., Aanensen D.M., Hanincová K., Terekhova D., Vollmer S.A., et. al. MLST of housekeeping genes captures geographic population structure and suggests a European origin of Borrelia burgdorferi. Proc. Natl Acad. Sci. USA. 2008; 105(25): 8730–5. https://doi.org/10.1073/pnas.0800323105
- Richter D., Postic D., Sertour N., Livey I., Matuschka F.R., Baranton G. Delineation of Borrelia burgdorferi sensu lato species by multilocus sequence analysis and confirmation of the delineation of Borrelia spielmanii sp. nov. Int. J. Syst. Evol. Microbiol. 2006; 56(Pt. 4): 873–81. https://doi.org/10.1099/ijs.0.64050-0
- Голидонова К.А., Коренберг Э.И., Крупинская Е.С. Сравнительный анализ результатов исследования изолятов боррелий методами мультилокусного сиквенс-анализа (MLSA) и типирования (MLST). Национальные приоритеты России. 2021; (3): 141–5.
- Нефедова В.В., Тетерин В.Ю., Коренберг Э.И., Горелова Н.Б., Воробьева Н.Н., Фризен В.И. Изоляция возбудителя иксодового клещевого боррелиоза из крови больных. Журнал микробиологии, эпидемиологии и иммунологии. 2009; (1): 63–6.
- Нефедова В.В., Коренберг Э.И., Горелова Н.Б. Генетические варианты Borrelia garinii — широко распространенного евразийского возбудителя заболеваний группы иксодовых клещевых боррелиозов. Молекулярная генетика, микробиология и вирусология. 2010; (3): 7–12.
- Голидонова К.А., Коренберг Э.И., Горелова Н.Б. Генетический анализ структуры некоторых генов домашнего хозяйства боррелий, вызывающих эритемную форму иксодового клещевого боррелиоза. В кн.: Сборник материалов VII Всероссийской междисциплинарной научно-практической конференции с международным участием «Социально-значимые и особо опасные инфекционные заболевания». Сочи; 2020: 49–50.
- Margos G., Vollmer S.A., Ogden N.H., Fish D. Population genetics, taxonomy, phylogeny and evolution of Borrelia burgdorferi sensu lato. Infect. Genet. Evol. 2011; 11(7): 1545–63. https://doi.org/10.1016/j.meegid.2011.07.022
- Casati S., Bernasconi M.V., Gern L., Piffaretti J.C. Diversity within Borrelia burgdorferi sensu lato genospecies in Switzerland by recA gene sequence. FEMS Microbiol. Lett. 2004; 238(1): 115–23. https://doi.org/10.1111/j.1574-6968.2004.tb09745.x
- Will G., Jauris-Heipke S., Schwab E., Busch U., Rossler D., Soutschek E., et al. Sequence analysis of ospA genes shows homogeneity within Borrelia burgdorferi sensu stricto and Borrelia afzelii strains but reveals major subgroups within the Borrelia garinii species. Med. Microbiol. Immunol. 1995; 184(2): 73–80. https://doi.org/10.1007/BF00221390
- Александров Г.О., Карпова М.Р., Потоцкая Ю.А., Бондаренко Е.И., Стронин О.В., Лукашова Л.В. Определение Borrelia miyamotoi — возбудителя клещевой возвратной лихорадки в клещах и крови пациента в Томской области. В кн.: Молекулярная диагностика – 2017: сборник трудов IХ Всероссийской научно-практической конференции с международным участием. М.; 2017: 338–9.
- Mantel N., Valand R.S. A technique of nonparametric multivariate analysis. Biometrics. 1970; 26(3): 547–58.
Дополнительные файлы


